






































Adsorption
Tematyka Badawcza
MAIN RESEARCH TOPICS
Research interests of our group are focused on the meaning and influence of adsorption phenomena on properties of systems revealing significant importance in applied science and/or biomedical applications. We also focus on model systems which analysis helps to understand the influence of microscopic factors on the observed macroscopic properties. .
Current research topics:
Research on the conformation of glycosaminoglycans. Glycosaminoglucans (GAGs) are a broad and diverse group of natural polysaccharides that play various and important roles in the functioning of living organisms. The influence of chemical functionalization (sulfation of selected exocyclic groups) on the conformation of the glycosidic bonds building the glycosaminoglycan chain was systematically investigated. Due to the strong structural heterogeneity and potential problems with the selection of representative structures, an alternative approach was used and the full set of 106 disaccharides that are present in molecules GAGs was considered. The aims of the research are: to find patterns of substitution that strongly disturb the geometry of the chain for a given type of compound, to explain the nature of mechanisms controlling chain conformation and to generate the data necessary for the next stage of research, i.e. the parameterization of GAG molecule for molecular dynamics simulation at the coarse-grained level of resolution.
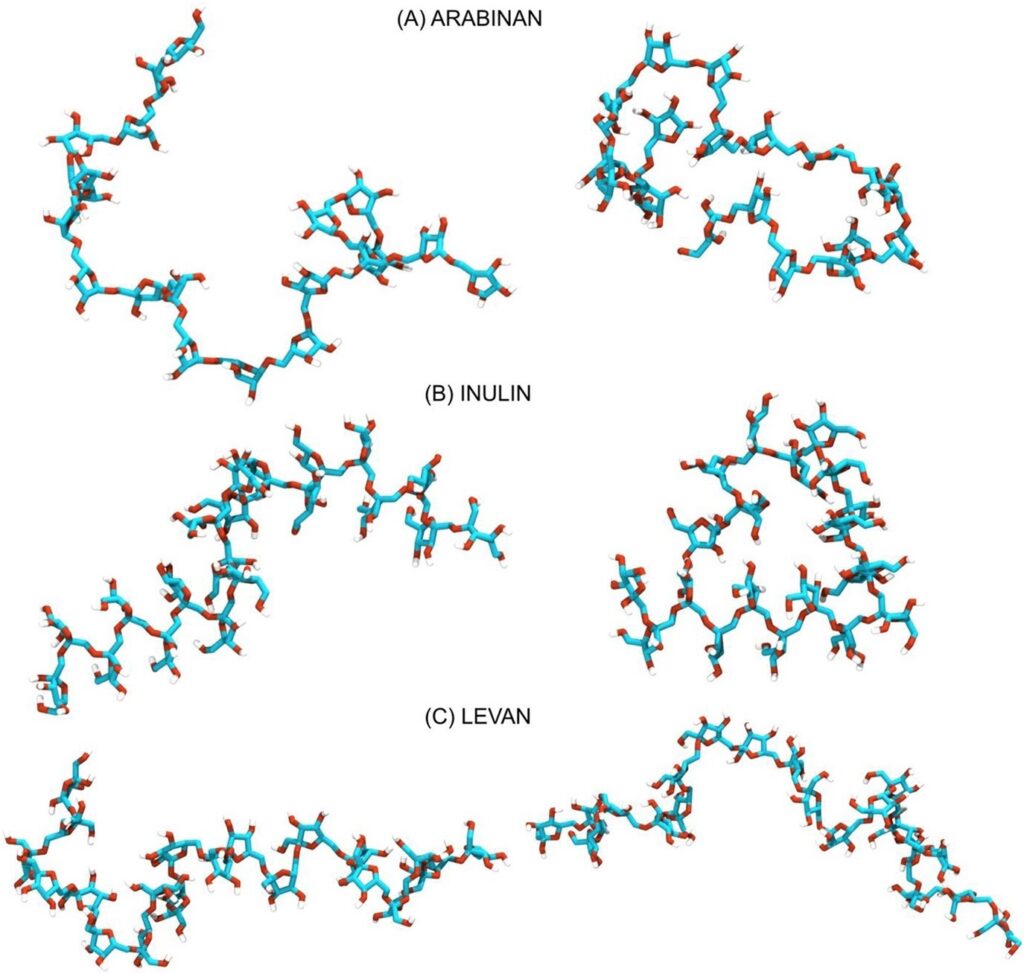
Fig. 1. Exemplary conformations of inulin, levan and arabinan chains generated as a result of molecular dynamics simulation with the use of a newly developed force field.
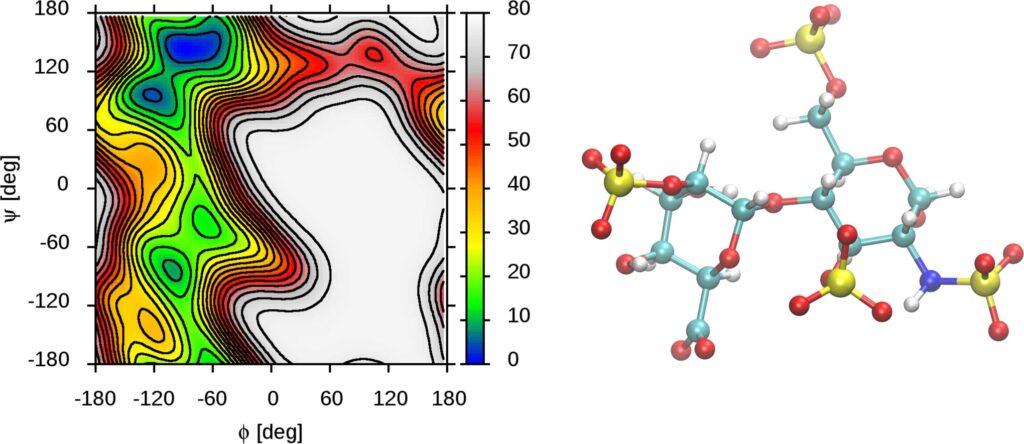
Fig. 2. Free energy map showing conformational properties of one of the disaccharides that make up the heparin sulphate chain.
Another branch of the research is development of a set of parameters (force field) designed to simulate a wide range of compounds belonging to the family of natural saccharides (including glycosaminoglycans). We aim to propose, test and validate coarse-grained parameters that are compatible with existing parameters for other types of biomolecules (with e.g. the MARTINI 3.0 set). The outcome will result in the ability to study the saccharide-containing biomolecular systems characterized by large dimensions and within microsecond timescales.
One of the research topics is the analysis of the structure of telomeric DNA fragments using molecular dynamics. Telomeres are the end parts of chromosomes made up of repetitive sequences of nitrogen bases (TTAGGG) 🙁 CCCTAA). Their role is to protect DNA against uncontrolled fusion during replication as well as to control the lifetime of the cell. Within telomeric DNA, both guanine and cytosine-rich strands can form higher-order spatial non-canonical structures: G-quadruplex and i-motif. The formation of these structures has a proven impact on the processes of blocking the infinite proliferative capacity of cells. The aim of the research is to analyze the processes of formation, deterioration and stability of G-quadruplex and i-motif structures depending on various factors, such as changing the pH of the environment or interaction with nanostructured objects. In particular, the adsorption processes of G-quadruplex and i-motif on the surface of carbon nanotubes are investigated. The analysis of these processes is of fundamental importance for the design of the so-called smart drug carriers.
The SEGO calculations are performed to study the mechanism / course of (i) the isomerization and fragmentation reactions, (ii) the intramolecular proton / methyl transfer induced by the external forces. We are also focusing on developing a new methodology for the use of external forces applied to atomic nuclei in molecules to locate transition states (TS) on the potential energy surface (PES). It is possible to move over PES from one transition state to another without involving any local minima (they can be located in a separate step through the reaction path calculations performed for every transition state found before). The proposed method is basically a reversal of the common PES search practice, when you can firstly locate the local minima, and then predict the structure of transition states connecting them.
One of the undertaken research tasks is the development of new classical, molecular mechanics force fields designed for studying the structure and conformation of carbohydrates and their subsequent use in the elucidate certain details related to the behaviors of these compounds. As a part of this project, we created the extensions of the GROMOS force field dedicated to the molecular dynamics simulations of the following classes of compounds: (i) carbohydrates containing ionized, protonated or esterified carboxyl groups (uronates, in particular: pectins and alginates); (ii) carbohydrates containing furanose rings (furanose monomers, e.g. ribose and arabinose; dimers, e.g. sucrose and oligomers, so-called fructans). In addition to the development, testing and validation of the newly designed force fields, a series of simulations conducted with the use of final parameters provided insight into a number of phenomena taking place at the molecular level with the participation of tested compounds.
Another field of research involves the use quantum mechanical methods to clarify some aspects of: (i) stereoelectronic effects associated with furanose molecules; (ii) anomeric, tautomeric and conformational equilibrium in mono-, di- and oligosaccharide molecules containing both pyranose and furanose units.
Dendrimers as a platform for designing biologically active carrier. Dendrimers are nanosized polymer systems with precisely controlled size, shape, molecular weight, composition, and reactivity. Two generations of positively charged poly(amidoamine) dendrimers (PAMAMs) were selected for the study as potential carriers for 5-fluorouracil (5FU), a drug primarily used in the treatment of colorectal cancer. Analytical techniques such as UV-Vis and NMR spectroscopy have shown that the most critical factor determining the formation of a PAMAM-5FU complex is the starting components’ protonation degree. The change in the system’s protonation degree was monitored using the zeta potential measurement as a function of the system’s pH and ionic strength. As a result of the dendrimer molecule interaction with the drug, a systematic decrease in charge of the complex was observed, indicating the dendrimer molecule’s charge compensation. However, these measurements do not provide precise information about the location of 5FU molecules in dendrimers’ structure. The observed shifts in the H1 NMR spectrum indicate drug molecules’ presence inside dendrimers’ structure and on its surface.
Additionally, using the QCM-D method, the adsorption efficiency and the number of drug molecules immobilized in the dendrimer structure were determined. The tests confirmed the system’s ability to attach approximately 20 5FU molecules per one dendrimer molecule for the fourth generation dendrimer and approximately 25 molecules for the sixth generation dendrimer. Comparing these values with the nominal number of amine groups present in a dendrimer structure, a system yield of 16% for the fourth generation (G4PAMAM) and 5% for six generation (G6PAMAM) dendrimers were obtained. These results correlate well with the spectroscopic studies.
Cytotoxicity tests (resazurin reduction and MTS tests) showed no toxicity of dendrimers against mouse fibroblast cells (L929) and a significant decrease in cell viability in the case of four human malignant cell lines: malignant melanoma (A375), glioblastoma (SNB-19), prostate cancer (Du-145) and colon adenocarcinoma (HT-29) during incubation with PAMAM-5FU complexes. The presence of the PAMAM dendrimer in the complex showed a statistically significant effect on cancer cells in the case of two cell lines (A375 and Du-145). Overall, the conducted research confirmed that PAMAM dendrimers could be active carriers of 5-fluorouracil for biomedical applications in cancer treatment.
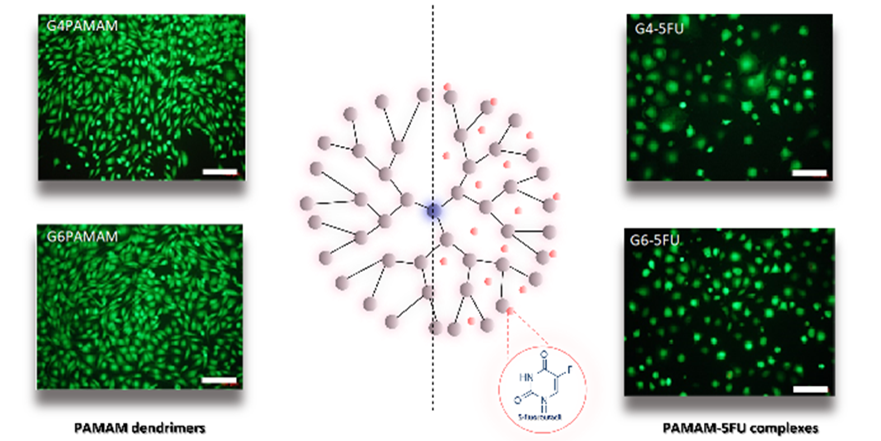
Fig. 3. The valuation of cytotoxicity of dendrimers PAMAM G 4.0 and G 6.0 and their complexes with 5FU against L929 mouse fibroblasts.
Understanding molecular aspects of the protein misfolding process: in situ spectroscopic and microscopic studies. Project research focuses on the characterization of protein layers at the solid/liquid interface. The structural stability and degree of hydration of the layers were monitored using a range of advanced analytical methods such as multi-parameter surface plasmon resonance (MP-SPR), quartz crystal microbalance (QCM-D), and infrared spectroscopy (FTIR). The SPR and QCM-D studies indicate that during the monolayer’s filling on the surface, lysozyme molecules’ orientation changes from side-on to between/end-on. In the range of high volumetric concentrations, bilayer formation is observed. The degree of hydration of the monolayers and bilayers differ significantly. The hydration degree for the monolayer varies between 35-75%, in the case of the bilayer is higher than 85%. Moreover, correlations between the degree of hydration of the adsorption layer and the layers’ viscoelastic properties were observed. In general, an increase in the filling of a layer is characterized by its stiffness. Simultaneously, a higher level of hydration of the system means an increase in the viscoelastic properties. Moreover, the protein’s interaction with the gold surface has a significant influence on the stability of the native structure of the protein. Based on FTIR spectra analysis, an increase in the content of β-turns and random structures of lysozyme is observed, with a simultaneous decrease in the range of β-sheets. The increase of β-turns in the structure determines the lysozyme stability and prevents system aggregation. The change in the system’s ionic strength induces a difference in the water shell structure around the protein molecule. Unbound water dominates at low salt content. The increase in salt concentration is associated with a more significant proportion of bound water, which affects the resulting layers’ stiffness and increases the stability of the II structure of the adsorbed protein
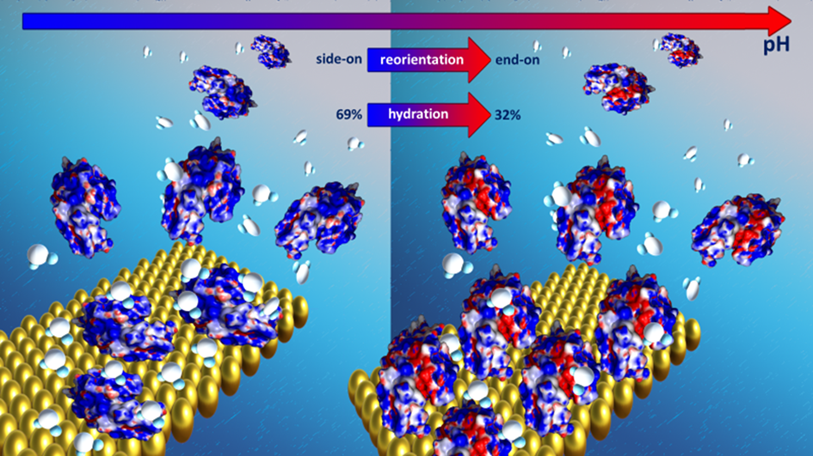
Fig. 4. Selfassembly and the degree of hydration of lysozyme layers on the Au surface.
Main achievements
The isolated form of the i-motif structure consists of a minimum of 22 nitrogen bases with the sequence (CCCTAA)3 Its existence is possible thanks to the formation of the so-called Hoogsteen pairs as opposed to the canonical form of DNA where the formation of complementary Watson-Crick pairs is responsible for the stability of the structure. I-motif shows significant thermodynamic stability in an acidic environment when Hoogsteen CC+ pairs can form between the protonated and the native form of cytosine. This property of i-motif has been exploited to construct a doxorubicin carrier that would be capable of releasing the drug in the acidic microenvironment of tumor tissue. Fig. 5 shows changes in the structure of the carbon nanotube, doxorubicin and ssDNA fragment with the sequence (CCCTAA)3CCCT depending on the pH. The analysis of these structures proves that doxorubicin and the ssDNA fragment adsorb stably on the surface of carbon nanotube at neutral pH, while at acidic pH the i-motif form is formed and doxorubicin molecules are released.
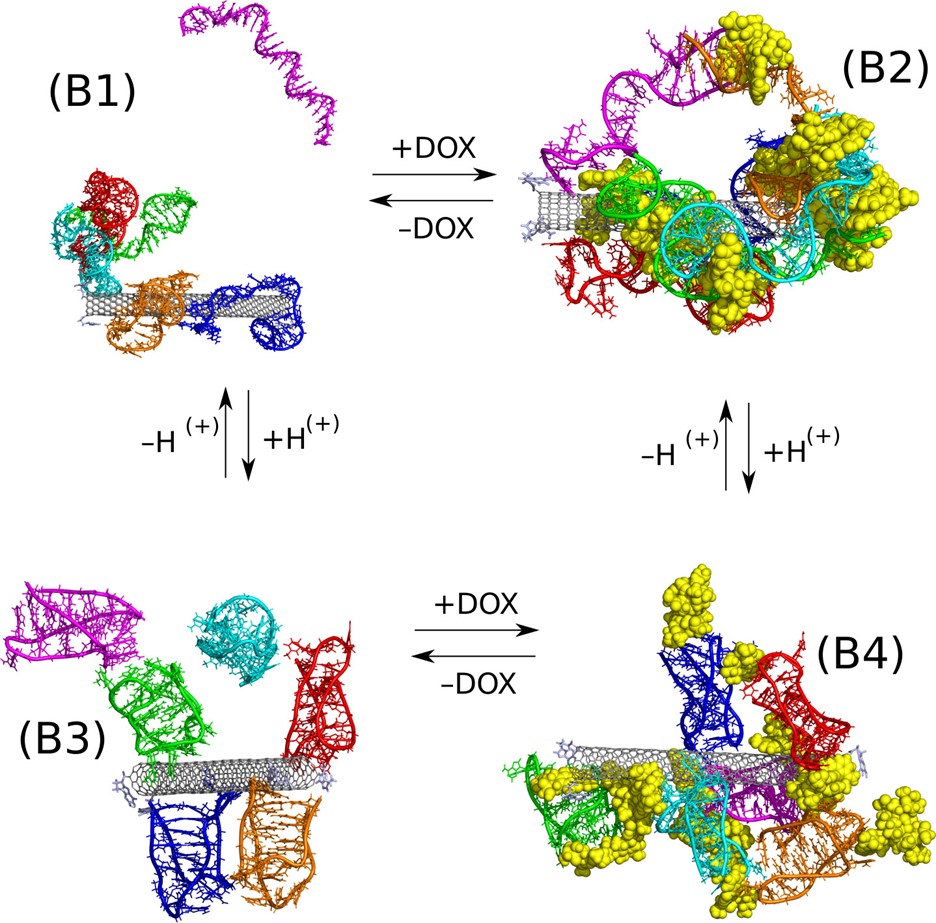
Fig. 5 Co-adsorption of doxorubicin and the DNA fragment with the sequence (CCCTAA)3CCCT having the ability to form i-motif at acidic pH.
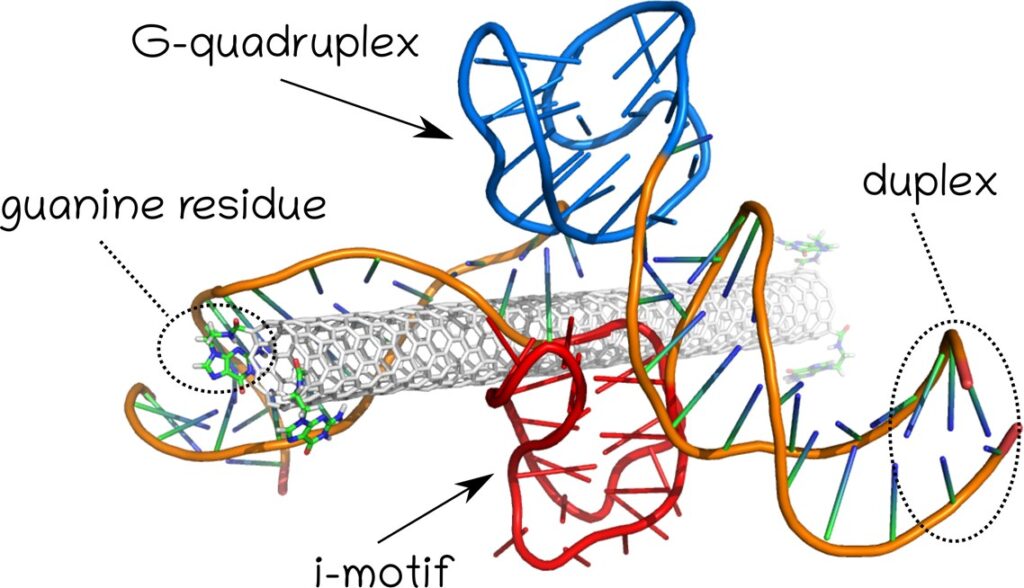
Fig. 6. Structure of the carbon nanotube, G-quadruplex and i-motif systems used to study the influence of the nanotube presence on the stability of non-canonical forms of DNA.
According to ScienceAlert, the structure of i-motif has recently (2018) been observed in a living cell, which proves that it can also occur under physiological conditions. Earlier studies of the structure of i-motif required a lowered pH and thus in vitro analyzes. As our calculations show, the existence of the i-motif at physiological pH may be related to the stabilizing role of the G-quadruplex formed in the immediate vicinity of the i-motif. However, detailed studies of the influence of the nanotube on the stability of i-motif in the system shown in Fig. 6 did not confirm the significant stabilization effect caused by the presence of the nanotube. Further studies on direct proton transfer from carboxyl groups attached to the carbon nanotube also did not confirm that the nanotube had the possibility of specific protonation of cytosines within i-motif DNA. Presumably, the key role is played by the dissociation of protons in the aqueous medium and the protonation of cytosines caused by the local decrease in pH.
The SEGO method (designed to automatically locate multiple minima on the molecular potential energy surface (PES)) was used to explore the potential energy surface of 2-methylfuran (2MF). This molecule recently gained a lot of interest as a potential biofuel candidate. Fig. 7 shows an exemplary isomerization and decomposition channel for 2MF obtained under the SEGO method.
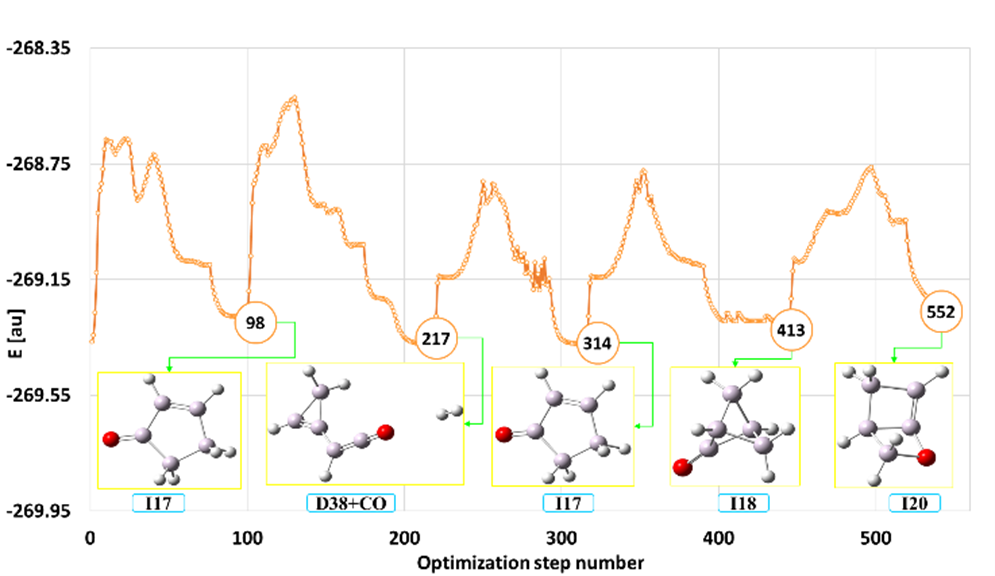
Fig. 7. Isomerization and decomposition channel for the 2-methylfuran molecule obtained by the SEGO method
Surprisingly, many local minima corresponding to 2MF isomers and fragmentation products (58 stable isomers and 40 different major degradation products) were located by the SEGO method. Some of the isomers and fragmentation products of 2MF have amazing structures that were probably previously unknown. They belong to different classes of chemical compounds with different functional groups and different bond systems. There are bi- and tricyclic compounds with both homo and hetero 3-, 4, 5 and 6-membered rings, chain structures and various functional groups (-OH, – C=O, -CHO, R-O-R, epoxy) combined with the presence of double and triple bonds between carbon atoms (alkene, alkyne, diene and allene). They can also be carbenes with one, two or even three divalent carbon atoms in the molecule. It should be emphasized that all structures were found automatically as products of forced chemical reactions controlled by the SEGO method.
A complementary part of the 2-methylfuran project was to investigate the mechanism of intramolecular proton / -CH3 transfer. This process is an initial step in 2MF pyrolysis reactions. Proton / methyl transfer was induced by specific external forces applied to the shifted protons or the carbon atom of the -CH3 group. The final proton transfer products were found automatically (Fig. 8) and all relevant transition states were located.
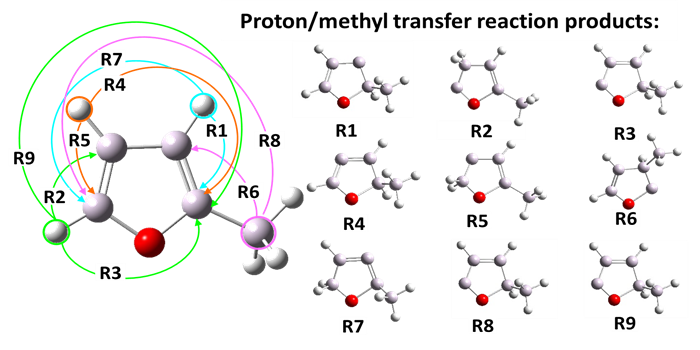
Fig. 8. The application of SEGO external forces to 2-methylfuran molecule and proton / CH3 transfer products.
A new methodology for exploring the potential energy surface using external forces has been proposed. In this way, a number of transition states can be located on the PES. Then, the corresponding local minima can be determined (by the standard reaction path calculations). Thus, this approach is, to some extent, a reversal method for determining stationary points on the PES. A number of transition states have been found for the 2-fluorofuran molecule. Fig. 9 presents the transition states (TS) and local minima (LM) found in fifteen independent one-step SEGO searches and their distances and energies relative to 2FF (LM0).
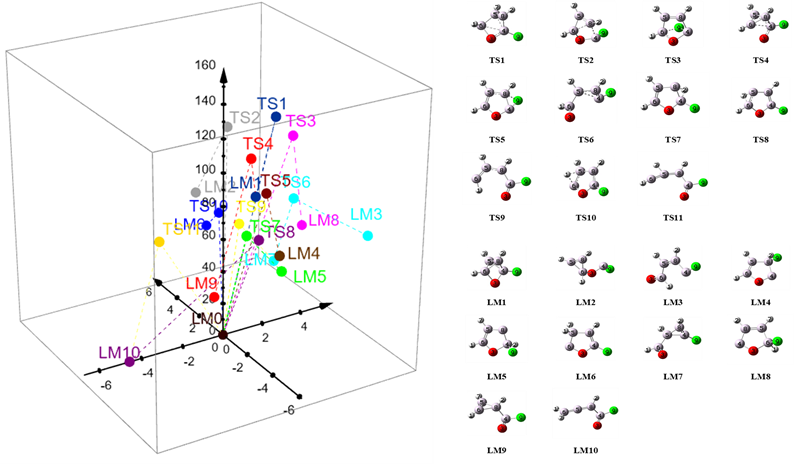
Fig.9. The transition states (TS) and the local minima (LM) found in fifteen independent one-step SEGO calculations and their energies (z-axis in kcal/mol) and distances (xy plane in Å) relative to 2-fluorofuran (LM0).
Development of the molecular mechanics force fields for simulation of carbohydrates containing either furanose or pyranose units and allowing for simulations of saccharides of any chain length (from the monomer level), with an arbitrary type of glycosidic bonds between monomers, varying anomery, chemical heterogeneity of the chain, possible functionalization of selected functional groups (O-alkylation, ionization or esterification of carboxylic groups) and possibility of branching. The developed force fields belong to the GROMOS family; they are compatible with the SPC water model and already existing parameter sets (from version 53A6 upwards, including the 56A6CARBO set for pyranoses).
Creation of a quantitative description of the effect of pH on conformational degrees of freedom in uronate molecules. The pH value affects the rotation of the exocyclic carboxylic substituent; depending on the considered compound, it also may affect the ring-inversion properties. The same variable slightly affects the conformation of the glycosidic linkage and does not affect the conformation of the lactol group.
Structural models of complexes created by pectin and alginate chains with calcium ions have been developed. It was shown that the dynamic conformations of complexes differing by the orientation of sugar chains (parallel vs. antiparallel) show a number of significant differences that can be interpreted in the context of calcium-induced aggregation occurring on a larger scale.
Systematic influence of the polar solvent on the conformation of the furanose ring has been proved to exist. This influence is manifested by generation of the energy barriers with a height of 3-7 kJ/mol, located at the OE and EO ring conformers. This effect is independent of the presence or absence of the ring substituent(s) and contributes to the wide applicability of the so-called two-state model, routinely used when analyzing the NMR data related to furanosides.
Finished research topics
Adsorption of magnetic nanoparticles on carbon nanotubes together with an analysis of their magnetically triggered desorption/detachment. Such processes are of great importance in designing of smart drug delivery systems being able to change their internal structure in response to some external triggering factor (eg. magnetic field). Thus, they are able to release encapsulated drugs in a given place and time. Therefore, a detailed analysis is devoted to interaction of typical therapeutic agents with carbon nanotubes as well as with various ligands which modify adsorptive properties of carbon nanotubes. Effects due to magnetic anisotropy and magnetization reversal within the nanoparticles body frames are also of great importance.
Theoretical study focused on the dynamics and thermodynamics of the conformational changes (1C4chair ↔ 4C1 chair) in molecules of the selected carbohydrates consisting of the six-membered pyranose rings.The ring conformation of the hexopyranose-based carbohydrate molecules is one of the central issues in glycobiology. The ring conformers (puckers) can determine the biological function and activity of carbohydrate and the dynamic equilibrium between puckers determines the macroscopic hydrodynamic properties of the carbohydrate polymers. Due to the extremely high free energy barriers separating particular ring conformers, most of the theoretical studies is focused on the detailed description of the free energy landscapes, neglecting the dynamic features of the conformational changes. Our study, based on the transition path sampling simulations is aimed at filling this gap.
The mutarotation of carbohydrates is one of the fundamental chemical reactions, important in the context various fields of Although being the classical ‘textbook’ reaction, some of its details still remain unknown. We plan to initiate the detailed computational investigation by using the molecular dynamics method, according to the QM/MM (quantum mechanics/molecular mechanics) protocol which means that part of the system (e.g. glucose molecule and selected, catalytic, water molecules) are modeled with the accuracy of quantum mechanics while the remaining part with that of classical biomolecular force fields (e.g. the TIP3P model of water)
Enforced structural changes in molecules. Theoretical studies are focused on various types of structural changes (e.g., conformational transitions, cis-trans isomerism, intramolecular rearrangements, etc.) caused by the external forces in the molecules of biological importance.
Adsorption of globular proteins on the biocompatible surface using a broad range of analytical techniques including quartz crystal microbalance with dissipation of energy (QCM-D) and surface plasmon resonance (SPR-MP). QCM-D and MP-SPR are powerful methods that enable highly sensitive, qualitative, real-time, label-free, and noninvasive detection of adsorbed proteins. Combinations of the above techniques have provided significant information on the mechanisms behind protein-material interactions, structural changes and biomolecular rearrangements. Work will be performed bidirectional, experimentally and theoretically with the use of molecular dynamics (MD). This project gives new perspectives for the development of novel protein/enzyme immobilization strategies for biomedical or biosensor applications.
Theoretical description of adsorption of simple ions and of surfactants at oxide/electrolyte interfaces (Special attention focused on enthalpies of adsorption and on effects of surface heterogeneity)
Equilibria and kinetics of gas adsorption on energetically heterogeneous solid surfaces, (also thermodesorption), adsorption in zeolites, and mixed-gas adsorption.
Main achievements in finished research topics
Design and computational study of the magnetically triggered molecular nanocontainer which is able to release of drug molecules (eg. cisplatin) in response to external magnetic field. Monte Carlo and Molecular Dynamics simulations allowed us to determine critical ranges of parameters which the nanocontiner must satisfy in order to work in the requested manner. The main factor responsible for the function of the nanocontineris its total energy profile accompanying the cycles of capping/uncapping. That energy is a function of dispersion forces between magnetic nanoparticle and carbon nanotube shell material which cover the magnetic core and magnetic properties of the core itself and sizes of both nanoparticle and carbon nanotube.The ground state of the nanocontainer is the capped state. The uncapping occurs as a result of the interaction with the external magnetic field, however, the magnetic nanoparticles must reveal high values of magnetic anisotropy constant. The release of drug (cisplatin) proceeds according to the activated one dimensional diffusion mechanism. It was found that that mechanism consists of two stages. A simple analytical formula which allows for prediction of long time limits of the release process was also derived.
In the investigation of the hexopyranose ring puckering we focused on the α-d- and β-d-glucopyranose molecules (GlcA and GlcB, respectively), treated as model systems. The results allowed for:
- identifying the distinct local minima of the free energy corresponding to the states intermediate for the 4C1↔ 1C4 transitions;
- assigning the time-characteristics to this transition and intermediate states;
- performing the search for the optimal reaction coordinate based on the Peters-Trout approach (likelihood maximization).
Additionally, the structures corresponding to the 4C1 ↔ 1C4 transition states (TS) have been found; surprisingly, in the case of GlcA, the water dynamics has very little influence on the probability of the TS evolution either to 4C1 or to 1C4. The different result obtained for GlcB (large influence of water dynamics on the behavior of TS as well as the poor applicability of the Peters-Trout approach for calculation of the reaction coordinate) speaks for slightly different mechanisms of the 4C1 ↔ 1C4 puckering in molecules of GlcB and GlcA, probably with the larger contribution of transitions over diffusive energy barriers, characteristic for rough free energy landscapes in the case of GlcA. The obtained results contribute to understanding the mechanisms governing the conformational changes in the carbohydrate rings. Additionally, the developed algorithms may be used to study other conformational changes in the related systems.
The atomic force microscopy (AFM) is a relevant tool to explore the mechanical/conformational stability of the biomolecules at single molecule level. The proper interpretation of experimental AFM force-extension curves is still difficult. Molecular insights on the origins of mechanical responses can be inferred from simulations and theoretical methods, e.g. the EGO model (Enforced Geometry Optimization). Experimental works dedicated to conformation changes in individual α -D-galacturonic acid molecules imposed by external forces showed that the transformation 4C1 → 1C4 takes place through the twisted boat conformation. The EGO simulation of the AFM experiment for α-D-galacturonic acid properly reproduces this experimental trend. The EGO model predicts essentially the same mechanism as the molecular dynamics and constrained geometry optimization calculations. The EGO method predicts three possible permanent conformational changes in the considered α-D-galacturonic acid oligomers (up to hexamer). The type of conformational transitions generally depends on the unit position in the oligosaccharide chain
Structure and properties of protein layers: from biomolecules to a functional layer (a combined experimental and simulation study). We performed analysis of conformational changes of lysozyme (LSZ) at various interfaces by Quartz Crystal Microbalance (QCM-D) and Surface Plasmon Resonance (MP-SPR). Furthermore, QCM-D can be used to study the adsorption behavior (kinetics, adsorbed amount and the thickness of protein film layer). Effect of solution concentration, pH and electrolyte type was studied to elucidate the nature of the association processes. We have found that the protein adsorption strongly depends on ionic strength and pH. MD simulation suggested that LSZ adsorbs strongly at surface through Arg and Lys residues from the N,C-terminal face. Moreover, the dominant interactions driving adsorption are the electrostatic interaction between LSZ and solid surface. Adsorbed LSZ can diffuse on the surface and create aggregates and clusters with other LSZ molecules.
Reversible swelling process of 6th generation poly (amidoamine) ( PAMAM) dendrimers. Our results supply the first reliable experimental evidence that swelling of G6 generation PAMAM dendrimer is reversible. Furthermore, we showed that this process is essentially modulated by condensation of the counterions at the dendrimer surface. These findings give a valuable insight into the dendrimer science and challenge for the further theoretical and experimental study, in particular, for assessing a degree of penetration of water and the counterions into dendrimer molecules, under the various conditions of pH, the ionic strength and the electrolyte type. This results have also a practical merit, because, they indicate that dendrimers may be used as stimulus-responsive drug delivery carriers, which can release a trapped drug upon a conformational transition induced in the dendrimer molecule by change in pH or the ionic strength.
Quantitative description of the enthalpic effects accompanying adsorption of simple ions at oxide/electrolyte interfaces. It includes all kinds of experiments carried out so far, i.e. batch experiments, calorimetric titration and flow calorimetry. Also studies of temperature dependence of ion adsorption were presented. This theoretical description was first developed for some model, well-defined surfaces of oxides, and next generalized by considering the energetic heterogeneity of the actual oxide/electrolyte interfaces. For the first time, it was shown, that drawing mathematical consequences of the existence of the Common Intersection Point makes it possible to establish interrelations of certain important physicochemical parameters.
Application of the Scaled Particle Theory to quantitatively describe the adsorption of surfactants at both solid/liquid and gas/liquid interfaces. That new theoretical description is at present the only one which has made possible a simultaneous quantitative description of both adsorption isotherms and of the accompanying enthalpic effects. It has been demonstrated that in the case of large surface aggregates the lateral interactions between these aggregates do not play a significant role in surfactant adsorption.
Theoretical description of the kinetics of gas adsorption on energetically heterogeneous solid surfaces. That new description was based on the Statistical Rate Theory of Interfacial Transport