





































Adsorpcja
Tematyka Badawcza
GŁÓWNE TEMATY BADAŃ
Zainteresowania badawcze grupy koncentrują się wokół znaczenia i wpływu zjawiska adsorpcji na właściwości układów o istotnym potencjale aplikacyjnym lub/i biomedycznym. Jak również układów modelowych pozwalających na zrozumienie wpływu fundamentalnych czynników mikroskopowych na obserwowane właściwości makroskopowe.
Aktualne badania prowadzone w grupie:
Badania nad konformacją glikozaminoglikanów (GAGs), tj. szeroką grupą naturalnych polisacharydów pełniących zróżnicowane i istotne role w funkcjonowaniu organizmów żywych. W sposób systematyczny zbadano wpływ funkcjonalizacji (sulfonowanie wybranych grup egzocyklicznych) na konformację wiązań glikozydowych budujących łańcuch glikozaminoglikanów. Z uwagi na silną heterogeniczność strukturalną oraz potencjalne problemy z wyborem reprezentatywnych struktur, zastosowano podejście skupiające się na pełnym zbiorze 106 disacharydów jakie mogą budować cząsteczki GAGs. Celem badań jest znalezienie wzorców podstawienia szczególnie silnie zaburzających geometrię łańcucha dla danego typu związku, wyjaśnienie ogólnych mechanizmów kontrolujących konformację łańcucha oraz wygenerowanie danych niezbędnych do kolejnego etapu badań, tj. parametryzacji cząsteczek GAGs do celów symulacji metodą dynamiki molekularnej na poziomie rozdzielczości coarse-grained. Kolejnym aspektem badań jest stworzenie zestawu parametrów (pola siłowego) do symulacji szerokiego zakresu związków z rodziny naturalnych sacharydów (włączając w to te należące do rodziny glikozaminoglikanów). Zamierzamy zaproponować, przetestować i zwalidować parametry typu coarse-grained, kompatybilne z istniejącymi parametrami dotyczącemi biocząsteczek innego typu (np. MARTINI 3.0). Efektem będzie możliwość badania układów biomolekularnych zawierających sacharydy i obejmujących znaczne rozmiary i dużą skalę czasową.
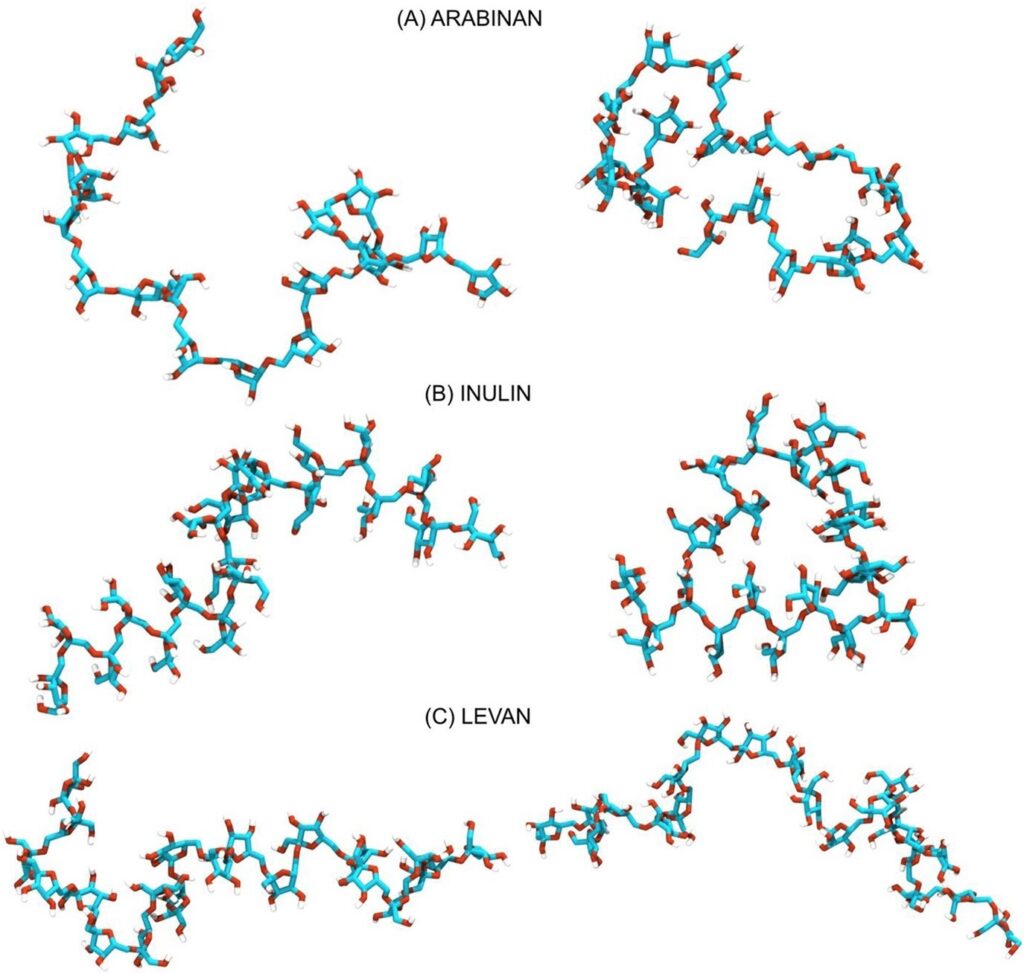
Rys. 1. Przykładowe konformacje łańcuchów inuliny, lewanu i arabinanu wygenerowane w wyniku symulacji metodą dynamiki molekularnej przy użyciu nowoopracowanego pola siłowego.
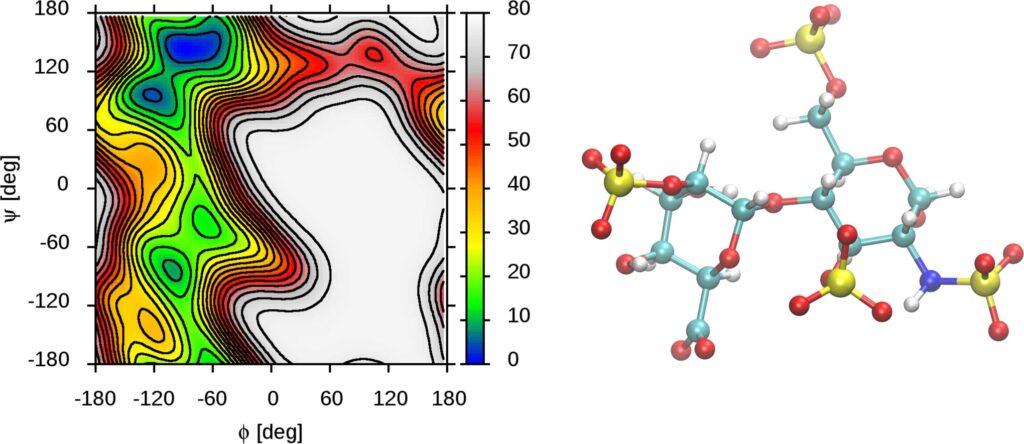
Rys. 2. Mapa energii swobodnej obrazująca właściwości konformacyjne jednego z disacharydów budujących łańcuch siarczanu heparyny.
Jednym z tematów badawczych jest analiza struktury telomerycznych fragmentów DNA przy użyciu dynamiki molekularnej. Telomery są to krańcowe fragmenty chromosomów zbudowane z wielokrotnie powtarzających się sekwencji zasad azotowych (TTAGGG):(CCCTAA). Ich rolą jest zabezpieczenie DNA przed niekontrolowaną fuzją podczas replikacji jak również kontrolowanie czasu życia komórki. W obrębie telomerycznego DNA zarówno te bogate w guaninę jak i cytozynę nici mogą tworzyć przestrzenne niekanoniczne struktury wyższego rzędu: G-quadruplex oraz i-motif. Formowanie się tych struktur ma udowodniony wpływ na procesy blokowania nieskończonego potencjału replikacyjnego komórek. Celem badań jest analiza procesów formowania się, rozpadu oraz stabilności struktur G-quadruplex oraz i-motif w zależności od różnych czynników jak np. zmiana pH środowiska czy oddziaływanie z obiektami nanostrukturalnymi. W szczególności badaniom poddawane są procesy adsorpcji G-quadruplexu oraz i-motifu na powierzchni nanorurek węglowych. Analiza tych procesów ma fundamentalne znaczenie dla projektowania tzw. inteligentnych nośników leków.
Prowadzone są obliczenia SEGO w celu badania mechanizmu/przebiegu wymuszonych siłami zewnętrznymi reakcji izomeryzacji i fragmentacji oraz wewnątrzcząsteczkowego transferu protonu/grupy metylowej. Badania skupiają się również na opracowaniu nowej metodologii dotyczącej wykorzystania zewnętrznych sił przykładanych do jąder atomowych w układzie molekularnym do lokalizacji stanów przejściowych (TS) na powierzchni energii potencjalnej (PES). Przechodzenie między kolejno znajdowanymi na PES stanami przejściowymi odbywa się bez jakiejkolwiek informacji o minimach lokalnych (można je oczywiście znaleźć w oddzielnym kroku w ramach obliczeń ścieżek reakcji). Proponowana metoda jest w zasadzie odwróceniem stosowanej dotychczas powszechnej praktyki przeszukiwania PES polegającej na lokalizacji minimów lokalnych, a następnie przewidywaniu struktury łączących je stanów przejściowych.
Jednym z zadań badawczych jest opracowywanie nowych klasycznych pól siłowych mechaniki molekularnej służących do badań struktury i konformacji węglowodanów oraz ich późniejsze wykorzystanie w badaniach tych związków. W ramach badań stworzyliśmy rozszerzenia pola siłowego GROMOS dedykowane do symulacji metodą dynamiki molekularnej następujących klas związków: (i) węglowodany zawierające zjonizowane, sprotonowane lub estryfikowane grupy karboksylowe (uroniany, w szczególności: pektyny oraz alginiany); (ii) węglowodany zawierające pierścienie furanozowe (monomery furanoz, np.: ryboza i arabinoza; dimery, np.: sukroza oraz oligomery, tzw. fruktany). Oprócz opracowywania, testowania oraz walidacji nowozaprojektowanych pól siłowych, symulacje przeprowadzone z użyciem finalnych parametrów oferują wgląd w szereg zjawisk zachodzących na poziomie molekularnym z udziałem badanych związków.
Prowadzone są również badania wykorzystujące metody chemii kwantowej w celu wyjaśnienia wybranych aspektów dotyczących: (i) efektów stereoelektronowych w cząsteczkach furanoz; (ii) równowagi anomerycznej, tautomerycznej oraz konformacyjnej w cząsteczkach mono-, di- i oligosacharydów zawierających zarówno jednostki piranozowe jak i furanozowe.
Dendrymery jako platforma do projektowania biologicznie czynnych nośników. Do badań wybrano dwie generacje dodatnio naładowanych dendrymerów poli (amidoaminy) (PAMAM) jako potencjalnych nośników dla 5- fluorouracyl, leku stosowanego głównie w leczeniu raka jelita grubego. Techniki analityczne, takie jak DLS, UV-Vis oraz LDV wykazały, że najważniejszym czynnikiem determinującym tworzenie kompleksu PAMAM-5FU jest stopień protonacji składników wyjściowych. Zmianę stopnia protonacji układu monitorowano stosując pomiar zeta potencjału w zależności od pH i siły jonowej układu. W wyniku oddziaływania cząsteczki dendrymeru z lekiem obserwowano systematyczny spadek ładunku kompleksu świadczący o kompensacji ładunku cząsteczki dendrymeru. Pomiary te nie dają jednak jednoznacznej informacji o lokalizacji cząsteczek 5FU w strukturze dendrymerów.
Dodatkowo stosując metodę QCM-D określono efektywność adsorpcji dendrymerów oraz wyznaczono liczbę cząsteczek leku immobilizowanych w strukturze dendrymeru. Badania potwierdziły zdolność układu do przyłączania około 20 cząsteczek 5FU na jedną cząsteczkę dendrymeru w przypadku czwartej generacji dendrymeru oraz około 25 cząsteczek w przypadku szóstej generacji dendrymeru. Porównując te wartości z nominalną liczbą grupy aminowych obecnych w strukturze dendrymeru, otrzymano wydajność układu na poziomie 16% dla G4PAMAM i 5% dla dendrymerów G6PAMAM. Wyniki te dobrze korelują z wynikami badań spektroskopowych.
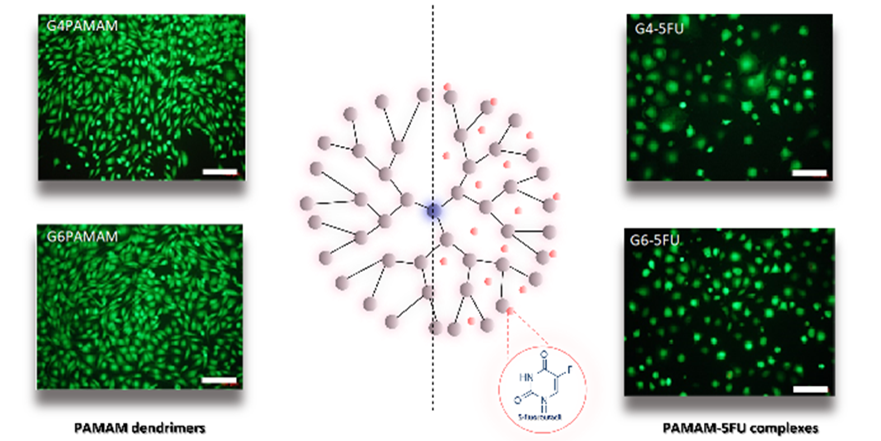
Rys. 3 Ocena cytotoksyczności dendrymerów PAMAM G 4.0 i G 6.0 oraz ich kompleksów z 5FU wobec mysich fibroblastów L929.
Zrozumienie molekularnych aspektów procesu nieprawidłowego fałdowania białek: badania in situ spektroskopowe i mikroskopowe. Prowadzone w ramach projektu badania skoncentrowane są na charakterystyce warstw białkowych na granicy międzyfazowej ciało stałe/ciecz. Stabilność strukturalna oraz stopień hydratacji warstw monitorowano stosując gamę zaawansowanych metod analitycznych takie jak wieloparametrowy powierzchniowy rezonans plazmonów (MP-SPR), mikrowagę kwarcową z dyssypacją energii (QCM-D) oraz spektroskopię w podczerwieni (FTIR). Badania SPR i QCM-D wskazują, iż w trakcie zapełniania monowarstwy na powierzchni mamy do czynienia ze zmianą orientacji cząsteczek lizozymu z pozycji side-on do between/end-on. W zakresie wysokich stężeń objętościowych obserwuje się tworzenie biwarstwy. Stopień hydratacji monowarstwy i biwarstwy różnią się znacząco. Stopień hydratacja dla monowastwy zmienia się w zakresie 35-75%, w przypadku biwarstwy to aż 85%. Ponadto zaobserwowano korelacje pomiędzy stopniem hydratacji warstwy adsorpcyjnej a własnościami lepkosprężystymi warstw. Generalnie wzrost zapełnienia warstwy charakteryzuje się zwiększeniem jej sztywności, natomiast wyższy poziom hydratacji układu to zwiększenie własności lepkosprężystych. Ponadto oddziaływanie białka z powierzchnią złota ma istotny wpływ na stabilność natywnej struktury białka. Na podstawie analizy widm FTIR obserwuje się wzrost zawartości struktur odpowiadających β-zwrotom oraz strukturom nieuporządkowanym lizozymu, przy jednoczesnym spadku zawartości β-kartek. Wzrost zawartości β-zwrotów w strukturze decyduje o stabilności struktury lizozymu i zapobiega agregacji układu. Zmiana siły jonowej układu indukuje zmianę struktury otoczki wodnej wokół cząsteczki białka. Przy niskiej zawartości soli dominuje woda nie związana. Wzrost stężenie soli wiąże się ze zwiększeniem udziału wody związanej wpływającej na sztywność powstałych warstw oraz stabilnością struktury II rzędowej adsorbowanego białka.
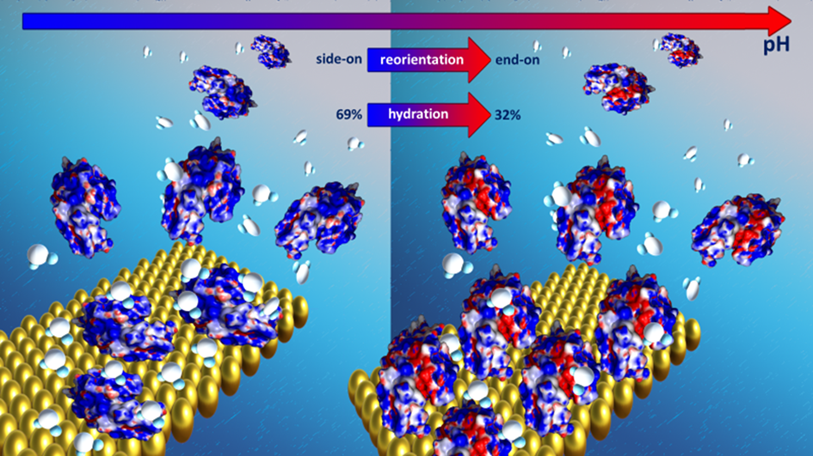
Rys. 4 Samoorganizacja oraz stopień hydratacji warstw lizozymu na powierzchni Au.
Główne osiągnięcia
Izolowana forma struktury i-motif składa się z minimum 22 zasad azotowych o sekwencji (CCCTAA)3 Jej istnienie jest możliwe dzięki tworzeniu się tzw. par Hoogsteena w odróżnieniu od kanonicznej formy DNA gdzie za stabilność struktury odpowiada tworzenie komplementarnych par Watsona-Cricka. I-motif wykazuje znaczą stabilność termodynamiczną w środowisku kwaśnym kiedy mogą tworzyć się pary Hoogsteena CC+pomiędzy sprotonowaną a natywną formą cytozyny. Ta właściwość i-motif została wykorzystana do konstrukcji nośnika doksorubicyny który byłby zdolny do uwalniania leku w kwaśnym mikrośrodowisku tkanki nowotworowej. Rys. 5 przedstawia zmiany struktury układu nanorurka węglowa, doksorubicyna oraz fragment ssDNA o sekwencji (CCCTAA)3CCCT w zależności od pH. Analiza tych struktur dowodzi, że doxorubicyna oraz fragment ssDNA adsorbują się stabilnie na powierzchni nanorurki węglowej w pH neutralnym, zaś w pH kwaśnym dochodzi do utworzenia formy i-motif oraz uswobodzenia cząsteczek doksorubicyny.
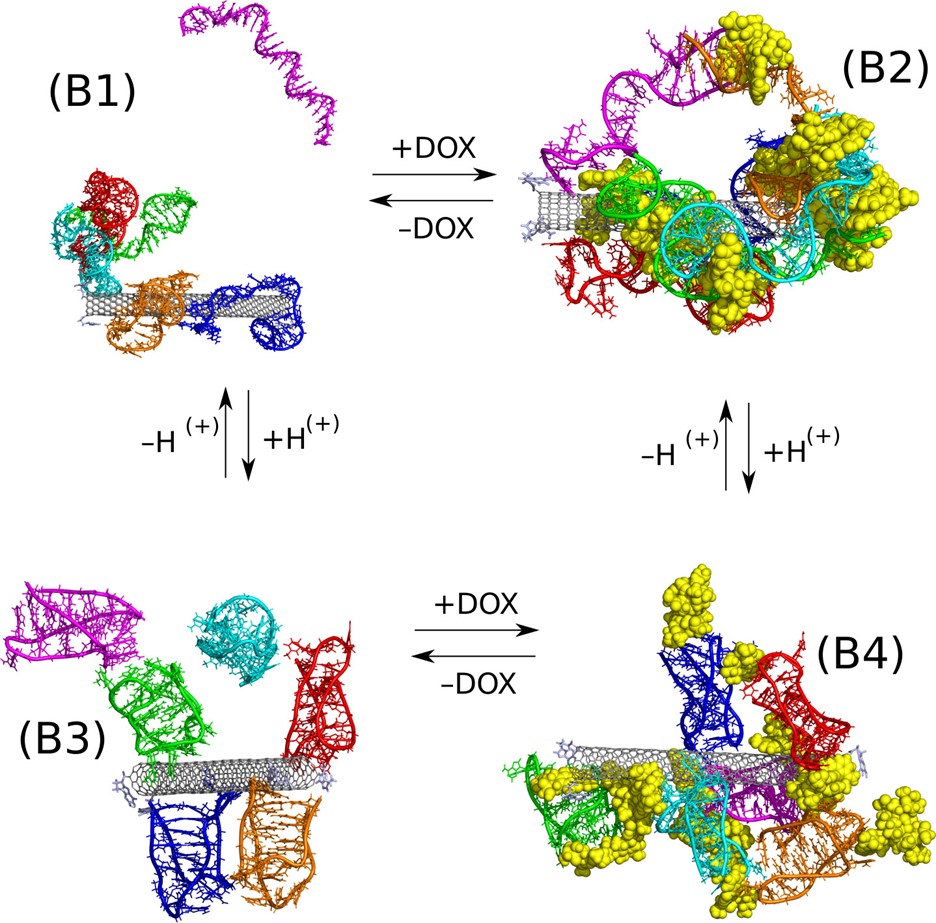
Rys.5 Koadsorpcja doksorubicyny oraz fragmentu DNA o sekwencji (CCCTAA)3CCCT mająca zdolność do tworzenia formy i-motif w pH kwaśnym.
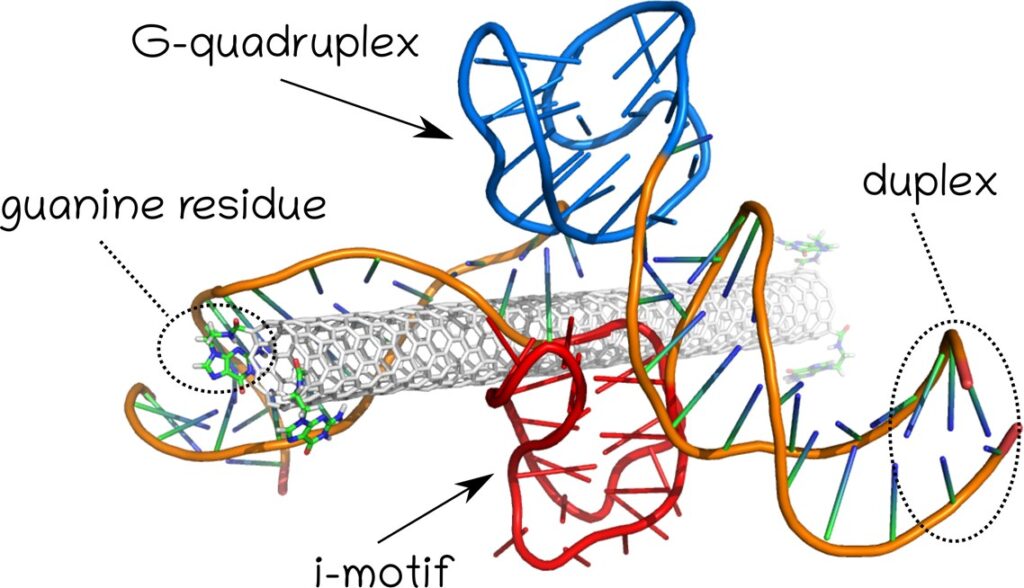
Rys. 6. Struktura układu nanorurka węglowa, G-quadruplex oraz i-motif stosowana do badania wpływu obecności nanorurki na stabilność niekanonicznych form DNA.
Jak podaje ScienceAlert struktura i-motif została niedawno (2018) zaobserwowana w żywej komórce co świadczy o tym, że może ona zaistnieć również w warunkach fizjologicznych. Wcześniejsze badania struktury i-motif wymagały obniżonego pH i tym samym analiz in vitro. Jak wynika z naszych obliczeń zaistnienie i-motifu w fizjologicznym pH może być związane ze stabilizującą rolą G-quadruplexu uformowanego w bezpośrednim sąsiedztwie i-motifu. Jednakże szczegółowe badania wpływu nanorurki na stabiliność i-motif w układzie przedstawionym na Rys. 6 nie potwierdziły istotnego efektu stabilizacji wywołanej obecnością nanorurki. Dalsze badania dotyczące bezpośredniego transferu protonu z grup karboksylowych przyłączonych do nanorurki węglowej również nie potwierdziły aby nanorurka miała możliwość specyficznej protonacji cytozyn w obrębie i-motif DNA. Przypuszczalnie kluczową rolę odrywa oddysocjowanie protonów w środowisku wodnym i protonacja cytozyn wywołana lokalnym obniżeniem pH.
SEGO (zaprojektowana z myślą o automatycznej lokalizacji wielokrotnych minimów na molekularnej powierzchni energii potencjalnej (PES)) została zastosowana do eksploracji powierzchni energii potencjalnej 2-metylofuranu (2MF). Molekuła ta budzi ostatnio duże zainteresowanie jako potencjalny kandydat na biopaliwo. Rys. 7 przedstawia przykładowy kanał izomeryzacji i dekompozycji dla 2MF uzyskany w ramach metody SEGO.
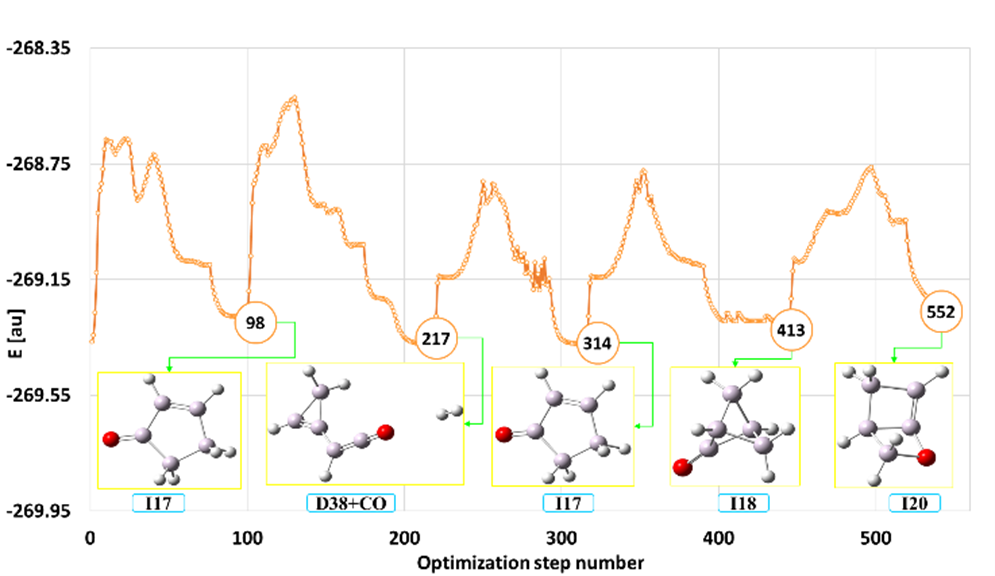
Rys. 7. Kanał izomeryzacji i dekompozycji dla cząsteczki 2-metylofuranu otrzymany w ramach metody SEGO
W przeprowadzonych badaniach zlokalizowanych zostało zaskakująco dużo lokalnych minimów odpowiadających izomerom 2MF i produktom fragmentacji (58 stabilnych izomerów i 40 różnych głównych produktów rozkładu). Niektóre izomery i produkty rozpadu 2MF mają zdumiewające struktury, które nie były najprawdopodobniej wcześniej znane. Należą one do różnych klas związków chemicznych z różnymi grupami funkcyjnymi i różnymi układami wiązań. Wyróżnić tu można związki bi- i tricykliczne o zarówno homo, jak i hetero 3-, 4, 5 i 6-członowych pierścieniach oraz strukturach łańcuchowych i różnorodnych grupach funkcyjnych (-OH, -C=O, -CHO, R-O-R, epoksydowej). Jednocześnie mogą się charakteryzować obecnością podwójnych i potrójnych wiązań między atomami węgla (alken, alkin, dien i allen). Mogą to również być karbeny z jednym, dwoma, a nawet trzema dwuwartościowymi atomy węgla w cząsteczce. Należy podkreślić, że wszystkie struktury zostały znalezione w sposób automatyczny w wyniku wymuszonych reakcji chemicznych sterowanych metodą SEGO.
Komplementarną częścią projektu dotyczącego 2-metylofuranu było zbadanie mechanizmu wewnątrzcząsteczkowego transferu protonu/grupy -CH3. Proces ten jest etapem początkowym w reakcjach pirolizy 2MF. Transfer protonu/grupy metylowej był indukowany przez określone siły zewnętrzne przyłożone do przesuwanych protonów lub atomu węgla z grupy -CH3. Pozwoliło to automatycznie znaleźć końcowe produkty transferu protonów (Rys. 8) i zlokalizować wszystkie odpowiednie stany przejściowe
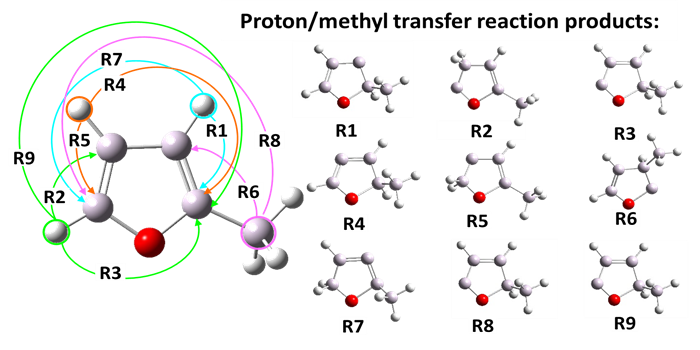
Rys. 8. Sposób przyłożenia sił zewnętrznych SEGO oraz produkty transferu protonu/grupy-CH3 w cząsteczce 2-metylofuranu.
Zaproponowana została nowa metodologia eksploracji powierzchni energii potencjalnej przy użyciu sił zewnętrznych. Podejście to pozwala na lokalizację na PES szeregu stanów przejściowych, a następnie w ramach obliczeń ścieżki reakcji – odpowiadających im minimów lokalnych. Jest więc w pewnym stopniu odwróceniem klasycznego sposobu wyznaczania punktów stacjonarnych na PES. Dla cząsteczki 2-fluorofuranu znalezionych zostało szereg stanów przejściowych. Diagram obrazujący wyniki: stany przejściowe (TS) i minima lokalne (LM) znalezione w piętnastu niezależnych jednoetapowych wyszukiwaniach SEGO oraz ich odległości i energie względem 2FF (LM0) przedstawiono na Rys. 9.
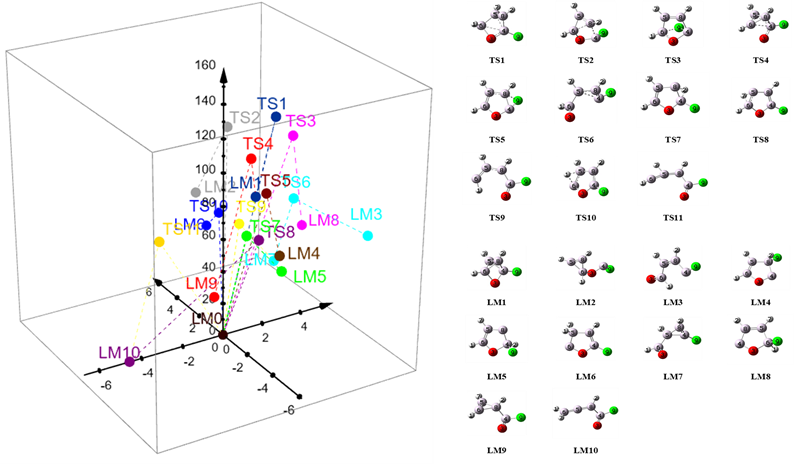
Rys. 9. Stany przejściowe (TS) i minima lokalne (LM) znalezione w piętnastu niezależnych jednoetapowych obliczeniach SEGO oraz ich energie (oś z w kcal/mol) i odległości (płaszczyzna xy w Å względem 2-fluorofuranu (LM0).
Opracowanie zestawów parametrów (pól siłowych) z przeznaczeniem do symulacji węglowodanów o jednostkach zarówno furanozowych jak i piranozowych, o dowolnej długości łańcucha (od poziomu monomeru), o arbitralnym typie wiązania glikozydowego pomiędzy monomerami, anomerii, heterogeniczości chemicznej łańcucha, możliwej funkcjonalizacji wybranych grup funkcyjnych (O-alkilacja, jonizacja lub estryfikacja grup karboksylowych) oraz rozgałęzień łańcucha. Opracowane pola siłowe należą do rodziny GROMOS, są kompatybilne z modelem wody SPC oraz istniejącymi już zestawami parametrów (od wersji 53A6 wzwyż, włącznie z zestawem 56A6CARBO dla piranoz).
Stworzenie ilościowego opisu wpływu pH na konformacyjne stopnie swobody w cząsteczkach uronianów. Wartość pH wpływa w stopniu znaczącym na rotację egzocyklicznego podstawnika karboksylowego, w zależności od rozpatrywanego związku może wpływać na właściwości inwersyjne pierścienia, w niewielkim stopni wpływa na konformację wiązania glikozydowego i nie wpływa na konformację grupy laktolowej.
Opracowano modele strukturalne kompleksów stworzonych przez łańcuchy pektyn oraz alginianów z jonami wapnia. Pokazano, że dynamiczne konformacje kompleksów różniących się orientacją łańcuchów cukrów (równoległa vs. antyrównoległa) wykazują szereg istotnych różnic, które mogą być interpretowane w kontekście agregacji zachodzącej w większej skali.
Udowodniono istnienie systematycznego wpływu polarnego rozpuszczalnika na konformację pierścienia furanozowego. Wpływ ten objawia się wygenerowaniem barier energetycznych o wysokości 3-7 kJ/mol zlokalizowanych na konformerach OE oraz EO. Efekt ten jest niezależny od obecności lub braku podstawników pierścienia i przyczynia się do szerokiej stosowalności tzw. two-state model, używanego przy analizie danych NMR.
Zakończone tematy badawcze
Adsorpcja nanocząstek magnetycznych na powierzchniach nanorurek węglowych wraz z analizą procesów magnetycznie wyzwalanej desorpcji/odrywania. Procesy te mają znaczenie w obszarze projektowania inteligentnych nośników leków które w odpowiedzi na czynnik wyzwalający (tj. zewnętrzne pole magnetyczne) zmieniają strukturę molekularną i są w stanie uwolnić cząsteczki leku w określonym miejscu i czasie. Tym samym szczegółowym badaniom poddawane są oddziaływania typowych cząsteczek terapeutycznych z nanorurkami węglowymi, różnego typu ligandami modyfikującymi właściwości adsorpcyjne nanorurek węglowych jak też i efekty związane z anizotropią magnetyczną i odwróceniem magnetyzacji nanocząstek magnetycznych.
Dynamika oraz termodynamika przejść konformacyjnych (typu krzesło 1C4 ↔ krzesło 4C1) w cząsteczkach wybranych cukrów, zbudowanych z sześcioczłonowych pierścieni (heksopiranoz). Konformacja pierścienia w cząsteczkach węglowodanów heksopiranozowych jest jednym z centralnych zagadnień glikobiologicznych. Konformery pierścieniowe determinują biologiczną funkcję i aktywność węglowodanów a dynamiczna równowaga pomiędzy poszczególnymi konformerami reguluje m.in. makroskopowe właściwości hydrodynamiczne policukrów. Z uwagi na ekstremalnie wysokie bariery energii swobodnej oddzielające stany odpowiadające poszczególnym konformerom, większość badań jest skupionych na szczegółowym opisie profili energii swobodnej, przy zaniedbaniu opisu cech dynamicznych procesu deformacji pierścienia. Nasze badania, oparte o zastosowanie metody transition path sampling mają na celu m.in. uzupełnienie tego braku.
Badanie reakcji mutarotacji węglowodanów, która jest jednym z fundamentalnych procesów, istotnych z punktu widzenia wielu różnych dziedzin chemii oraz biochemii. Pomimo bycia „podręcznikową” reakcją chemii organicznej, pewne szczegóły mutarotacji ciągle nie są w pełni poznane. W ramach projektu planujemy przeprowadzić szczegółowe badania teoretyczne, zgodnie z protokołem dynamiki molekularnej w oparciu o potencjały oddziaływań ab initio połączone z klasycznymi polami siłowymi (QM/MM, quantum mechanics/molecular mechanics), co oznacza, że część układu (cząsteczka glukozy oraz cząsteczki wody biorące bezpośredni udział w reakcji — katalizujące ją) modelowane są z dokładnością mechaniki kwantowej; pozostała część — klasycznej mechaniki molekularnej (model wody TIP3P dla reszty środowiska wodnego).
Wymuszone zmiany strukturalne w molekułach. Badania teoretyczne dotyczą różnego typu zmian (np. przejść konformacyjnych, izomerii cis-trans, przegrupowań wewnątrzcząsteczkowych itp.) wywołanych przez siły zewnętrzne w cząsteczkach o znaczeniu biologicznym.
Adsorpcja białek globularnych na powierzchniach biokompatybilnych. Analiza warstw adsorpcyjnych z zastosowaniem precyzyjnych metod bezznacznikowych takich jak mikrowaga kwarcowa z dyssypacją energii (QCM-D) oraz powierzchniowy rezonans plazmonów (MP-SPR). Wyznaczenie kinetyki adsorpcji, zmian konformacji, stopnia odwracalności procesu adsorpcji oraz stabilności warstw białkowych. Skorelowanie wyników eksperymentalnych z metodami teoretycznymi. Określenie mechanizmu adsorpcji białka z zastosowaniem pełno atomowej dynamiki molekularnej (MD), wyznaczenie najważniejszych aminokwasów rządzących adsorpcją oraz zaproponowanie mechanizmu dyfuzji na modelowej powierzchni (z podaniem barier energetycznych) wybranego białka.
Adsorpcja jonów prostych i jonów surfaktantów na granicach faz tlenki/roztwór elektrolitu (ze szczególnym uwzględnieniem entalpii adsorpcji i efektów niejednorodności energetycznej powierzchni),
Kinetyka adsorpcji i adsorpcja równowagowej gazów na energetycznie i geometrycznie niejednorodnych ciałach stałych (także termodesorpcji)
Adsorpcja w zeolitach i adsorpcja z mieszanin gazowych.
Główne osiągnięcia w zakończonych tematach badawczych
Opracowanie modelu magnetycznie sterowanego nanopojemnika molekularnego, który umożliwia kontrolowane dostarczenie i uwolnienie leku (cisplatyna). W oparciu o modelowanie z użyciem metod Monte Carlo i Dynamiki Molekularnej wyznaczono zakresy parametrów, w których nanopojemnik wykazuje pożądane właściwości. Głównym czynnikiem odpowiedzialnym za właściwe działanie nanopojemnika jest profil energii całkowitej towarzyszący procesowi zamykania/otwierania. Energia ta jest funkcją: oddziaływań dyspersyjnych pomiędzy nanocząstką magnetyczną a nanorurką, materiału powłoki nanocząstki magnetycznej i jej parameterów magnetycznych oraz rozmiaru nanocząstki i średnicy nanorurki. Stanem podstawowym nanopojemnika jest stan zamknięty. Otwarcie zachodzi wyłącznie wskutek oddziaływania z zewnętrznym polem magnetycznym, przy czym nanocząstki magnetyczne muszą wykazywać wysokie wartości bariery anizotropii magnetycznej. Uwalnianie leku (cisplatyny) zachodzi według mechanizmu aktywowanej dyfuzji jednowymiarowej. Wykazano, że jest to mechanizm dwuetapowy oraz wyprowadzono proste równanie analityczne pozwalające na przewidzenie szybkości uwalniania cisplatyny z wnętrza nanopojemnika dla długich czasów.
Badania zmian konformacyjnych w pierścieniach glukopiranoz skupione były na cząsteczkach α-D- i β-D-glukopiranozy (odpowiednio, GlcA i GlcB), zanurzonych w środowisku wodnym i traktowanych jako układy modelowe. Wyniki pozwoliły na:
- zidentyfikowanie odrębnych minimów lokalnych energii swobodnej, odpowiadających stanom przejściowym dla zmiany konformacyjnej 4C1↔ 1C4;
- przypisanie charakterystyki czasowej do ww. zmiany oraz stanów przejściowych;
- przeprowadzenie poszukiwań optymalnej współrzędnej reakcji 4C1 ↔ 1C4, opartych na procedurze Petersa-Trouta.
Dodatkowo, struktury odpowiadające stanowi przejściowemu reakcji 4C1 ↔ 1C4 (TS) zostały zidentyfikowane i poddane analizie. Wbrew oczekiwaniom, okazało się, że dynamika wody nie ma większego wpływu na prawdopodobieństwo ewolucji struktur będących TS dla GlcA do 4C1 lub 1C4. Odmienne rezultaty otrzymane dla GlcB (tj. znaczny wpływ dynamiki wody na zachowanie TS oraz słaba stosowalność procedury Petersa-Trouta) przemawiają za nieco innymi mechanizmami rządzącymi deformacją pierścieni GlcA i GlcB, prawdopodobnie z większym wkładem ‘dyfuzyjnego’ błądzenia układu po krajobrazie energii swobodnej w przypadku GlcA.
Wyniki stanowią istotny wkład do zrozumienia mechanizmów zmian konformacyjnych w pierścieniach węglowodanów na poziomie molekularnym. Ponadto, zmodyfikowane algorytmy TPS mogą zostać użyte do badań przejść konformacyjnych innego typu w zbliżonych właściwościami układach.
Mikroskopia sił atomowych (AFM) jest dogodną metodą do badania mechanicznej/konformacyjnej stabilności biomolekuł na poziomie pojedynczej cząsteczki. Właściwa interpretacja eksperymentalnych krzywych FC (ang. force-extension) pozostaje nadal zadaniem trudnym. Metody i symulacje teoretyczne (takie np. jak model EGO (ang. Enforced Geometry Optimization) mogą być pomocne w analizie właściwości mechanicznych cząsteczek. Badania eksperymentalne dotyczące zmian konformacyjnych wywołanych przez siły zewnętrzne w cząsteczkach kwasu α-D-galakturonowego wskazują, że przejście konformacyjne 4C1→ 1C4 zachodzi poprzez konformacje skręconej łódki. Symulacje EGO prawidłowo odtwarzają ten doświadczalny trend. Wyniki uzyskane w ramach modelu EGO są zasadniczo zgodne z opisem mechanizmu w/w przejścia konformacyjnego uzyskanym w ramach dynamiki molekularnej i obliczeń tzw. ograniczonej optymalizacji geometrii (ang. constrained geometry optimization). Model EGO przewiduje trzy możliwe trwałe przejścia konformacyjne w rozważanych oligomerach kwasu α-D-galakturonowego (do heksameru). Rodzaj przejścia konformacyjnego generalnie zależy od położenia danej jednostki (meru) w łańcuchu oligosacharydu.
Struktura i Właściwości Warstw Białkowych: od Biomolekuły do Funkcjonalnej Warstwy. Określono mechanizm adsorpcji lisozymu na powierzchni miki oraz krzemu. Przeprowadzono analizę zmian konformacji oraz reorganizacji struktury białka. Zastosowano precyzyjne metody charakterystyki objętościowej (DLS, lepkość dynamiczna, ruchliwość elektroforetyczna) do badań zachowania białka w roztworach wodnych. Do wyznaczenia struktury warstw białkowych tworzonych na powierzchniach biokompatybilnych wybrano mikrowagę kwarcową z dyssypacją energii (QCM-D) oraz powierzchniowy rezonans plaznonów (MP-SPR). Wyznaczono korelacji pomiędzy gęstością i strukturą warstw białkowych na powierzchniach adsorpcyjnych a własnościami białka w roztworze. Do modelowanie adsorpcji białka zastosowano metodę pełno atomowej dynamiki molekularnej (MD). Wytypowano najważniejsze aminokwasy, siły rządzące adsorpcja oraz zaproponowano mechanizm dyfuzji na powierzchni (z podaniem barier energetycznych).
Odwracalny Proces Pęcznienia Dendrymerów PAMAM Przeprowadzono badania dostarczające wiarygodnych dowodów eksperymentalnych, iż proces pęcznienia dendrymerów G6 PAMAM jest odwracalny. Ponadto, wykazano, że proces ten jest głównie regulowany przez kondensację przeciwjonów na powierzchni czasteczki dendrymeru. Ustalenia te mają cenny wgląd we własności fizykochemiczne dendrymerów i są wyzwaniem dla dalszych badań zarówno teoretycznych jak i eksperymentalnych, w szczególności, do oceny stopnia penetracji wody i przeciwjonów do cząsteczek dendrymeru, w różnych warunkach pH, siły jonowej oraz typu elektrolitu. Wyniki te maja również praktyczne znaczenie, ponieważ wskazują, że dendrymery mogą być stosowane jako nośniki leków. Stymulując konformacyjną zmianą indukowaną w cząsteczce dendrymeru przez zmianę pH lub siły jonowej, może uwalniać lek z cząsteczki nośnika.
Opracowano ilościowy opis efektów entalpowych towarzyszących procesowi adsorpcji jonów prostych na granicy faz tlenek/roztwór elektrolitu
W oparciu o Scaled Particle Theory opracowano ilościowy opis adsorpcji surfaktantów na granicy faz ciało stałe/ciecz oraz gaz/ciecz. Ten model teoretyczny jako jedyny umożliwiał jednoczesny ilościowy opis izoterm adsorpcji i towarzyszących efektów entalpowych.
W oparciu o Statystyczną Teorię Transportu (SRT) opracowano modele kinetyki adsorpcji gazów na energetycznie niejednorodnych powierzchniach ciał stałych. Udowodniono, że szereg równań empirycznych stosowanych do kinetyki adsorpcji może być bezpośrednio wyprowadzony na bazie Statystycznej Teorii Transportu. Wykazano też, że złożone zależności współczynników przyklejenia w funkcji stopnia pokrycia mogą być wyjaśnione w oparciu o SRT bez konieczności wprowadzania koncepcji tzw. precursor states.
Wykazano, w oparciu o teorię fraktalną, że istnieje związek pomiędzy niejednorodnością energetyczną powierzchni a jej strukturą geometryczną. Pozwala to na zdefiniowanie prostych zależności pomiędzy funkcją rozkładu energii adsorpcji a rozkładem objętości porów.