




































Enzymy – katalizatory życia
Maciej Szaleniec*, Agnieszka M. Wojtkiewicz (Rugor)
Instytut Katalizy Fizykochemii Powierzchni im. Jerzego Habera, Polskiej Akademii Nauk, Niezapominajek 8, 30-239 Kraków, *maciej.szaleniec@ikifp.edu.pl
Abstrakt
Enzymy to białkowe katalizatory warunkujące zachodzenie niemal wszystkich ważniejszych procesów życiowych. Dzięki ich niesamowitej, wciąż nie do końca wyjaśnionej katalitycznej skuteczności, skomplikowane przemiany chemiczne w naszym organizmie mogą zachodzić w bardzo łagodnych warunkach (niskie temperatury, niewysokie ciśnienia), w porównaniu do tych, jakie stosowane są w procesach przemysłowych. Naukowy dowód, że mogą one działać również poza organizmami żywymi, ostatecznie pogrzebał teorię panwitalizmu i zapoczątkował bardzo burzliwy rozwój nowoczesnej enzymologii i biotechnologii. Dzięki zaś rozwojowi technik biologii molekularnej i inżynierii genetycznej enzymy znajdują coraz szersze zastosowanie. Niepostrzeżenie w ciągu ostatnich kilku dekad enzymy stały się niezbędnym narzędziem w przemyśle farmaceutycznym, spożywczym, skórzanym, tekstylnym, papierniczym a nawet w proszkach do prania. Ułatwiają pracę naszych komórek, sprawiają, że jemy smaczniejszy chleb i pijemy słodki sok pomarańczowy nawet w środku zimy, a nasze ubrania są czyste już po praniu w 30oC.
Enzym – biologiczny katalizator
Enzymy należą do substancji, które są przez chemików nazywane katalizatorami. Każdy słyszał o katalizatorach zainstalowanych w samochodach, ale sam termin może czasem wydawać się niejasny. Jeżeli zapytamy chemika czym jest katalizator, odpowie, zgodnie z dość hermetyczną definicją, że jest to substancja, która przyspiesza reakcję chemiczną i skierowuje ją na wybraną ścieżkę, sama nie zużywając się w reakcji. Aby zrozumieć o co w tym wszystkim chodzi wyobraźmy sobie reakcję chemiczną, na przykład zachodzącą w komórkach czy w układzie wydechowym samochodu, jako zatłoczone rondo w środku dużego miasta. Samochody wjeżdżające drogą na rondo to substancje chemiczne, które będą ulegały przekształceniu (tzw. substraty), zaś każda z dróg opuszczająca rondo to inna ścieżka reakcji. Z kolei samochody opuszczające rondo to produkty, różniące się pod względem budowy cząsteczki chemiczne. Ponieważ rondo jest zakorkowane, wszystkie reakcje postępują dość wolno i każdy samochód wjeżdżający na nie straci sporo czasu zanim je opuści (tj. przemieni się w produkt reakcji). Katalizator możemy wyobrazić sobie jako wielką estakadę, przerzuconą nad zatłoczonym rondem, która wiedzie w kierunku najbardziej potrzebnym dla naszej komórki czy środowiska. Teraz samochody bardzo szybko przemykają nad korkiem (przyspieszenie reakcji) i większość z nich wybierze właśnie tę drogę (skierowanie ruchu na wybraną ścieżkę). Dokładnie według tej prostej analogii działa zarówno katalizator w samochodzie, odpowiednio utleniający resztki benzyny i redukujący tlenki azotu w spalinach, jak i enzym w naszym jelicie ułatwiający trawienie pożywienia.

Ryc. 1. Energia aktywacji (tj. różnica energii stanu przejściowego (S#) i energii substratów) ulega zmniejszeniu na skutek katalitycznego działania enzymu (różnica energii ES# i substratów). Obniżenie energii aktywacji reakcji katalizowanej w porównaniu do niekatalizowanej odpowiada za przyspieszenie reakcji przez katalizator.
Efekt estakady nad korkiem jest nazywany przez chemików „obniżeniem energii aktywacji” (Ryc. 1). Zgodnie z klasyczną teorią stanu przejściowego, każdą reakcję chemiczną charakteryzuje pewna określona energia aktywacji – im jest ona wyższa, tym dłużej musimy poczekać, aby reakcja miała miejsce. Zastanówmy się skąd bierze się wspomniana wyżej energia aktywacji. Otóż na co dzień w przeważającym stopniu cząsteczki chemiczne pozostają w niskich stanach energetycznych. Aby doszło do reakcji chemicznej pomiędzy cząsteczkami muszą one najpierw zbliżyć się do siebie, pokonując odpychające działania elektrostatyczne i ulegając przy tym niekorzystnym energetycznie odkształceniom związanym z reakcją chemiczną. Na przykład w czasie przeniesienia atomu z jednej cząsteczki do drugiej w tak zwanym stanie przejściowym następuje rozciągnięcie wiązania, które ulega odkształceniu. Te zmiany geometrii i struktury elektronowej związane są właśnie z podniesieniem energii układu, którą nazywamy energią aktywacji. Jeżeli taka bariera energetyczna jest wysoka, tylko niewielka ilość cząsteczek ma wystarczającą energię, aby ją pokonać. W rezultacie tylko nieliczne molekuły, z olbrzymiej liczby cząsteczek, które potencjalnie mogłyby zareagować, są zdolne do chemicznej przemiany, a co za tym idzie sama reakcja (z naszego makroskopowego punktu widzenia) postępuje bardzo powoli (tzn. mało produktu powstaje w czasie). Aby wpłynąć na szybkość reakcji możemy zwiększyć energię cząsteczek podnosząc ich temperaturę. Wraz ze wzrostem temperatury coraz więcej cząsteczek, zgodnie z rozkładem Boltzmana (Ryc. 2), osiąga wyższe energie i tym samym będzie zdolnych osiągnąć i skutecznie przekroczyć barierę reakcji. W rezultacie więcej cząsteczek w danym czasie jest w stanie przemienić się w produkt, co skutkuje eksponencjalnym wzrostem obserwowanej szybkości reakcji [1, 4]. Podgrzewanie reagentów jest jednak procesem bardzo kosztownym, a czasem, gdy mamy do czynienia z wrażliwymi termicznie związkami chemicznymi o skomplikowanej strukturze, nawet zupełnie niewykonalnym. W takiej sytuacji jedynym możliwym sposobem jest zastosowanie katalizatora.

Ryc. 2. Rozkład energii kinetycznej cząsteczek w zależności od temperatury układu wg. rozkładu Boltzmana. W miarę podnoszenia temperatury coraz większa populacja cząsteczek osiąga wymaganą granicę energii aktywacji, co jest warunkiem koniecznym do zajścia reakcji chemicznej. Na zielono zaznaczono część populacji cząsteczek, które w temperaturze 200K osiągają przynajmniej energię równą energii aktywacji.
Rolą katalizatora jest tak wpłynąć na cząsteczki w stanie przejściowym, aby ich energia nie musiała być aż tak wysoka by reakcja zachodziła (tj. obniżyć energię aktywacji). W rezultacie, podobnie jak w przypadku podniesienia temperatury, większa ilość cząsteczek chemicznych ma szansę pokonać barierę i reakcja przebiega szybciej. Takimi katalizatorami są właśnie enzymy, które do perfekcji realizują to właśnie zadanie. Wiążą one reagenty w swoim wnętrzu w taki sposób, aby maksymalnie obniżyć energię ich aktywacji [15] i przyspieszyć proces tworzenia produktu.
Bariery energetycznej, w metaforycznym sensie, doświadcza każdy z nas po niezbyt dobrze przespanej nocy, gdy stara się rano wstać do pracy – W tym przypadku katalizator, na przykład w postaci zapachu dobrej mocnej kawy pomaga znacząco obniżyć barierę energii aktywacji i pomóc nam wstać.
Precyzyjna regulacja
Enzymy są niezwykłymi katalizatorami. Dzięki swojej dość skomplikowanej i wyrafinowanej budowie potrafią działać tylko w określonym miejscu naszego organizmu i w określonych warunkach temperatury i pH. Są w stanie również rozpoznawać cząsteczki chemiczne i spośród wielu obecnych w naszych komórkach wybierać tylko te, do których modyfikacji zostały przeznaczone. Zjawisko to, nazywane specyficznością enzymów, może powodować że enzymy wybierają wyłącznie jeden rodzaj cząsteczki (np. absolutna specyficzność enzymu laktazy, który katalizuje rozkład wyłącznie laktozy) bądź też jakąś ich grupę (specyficzność grupowa – np. enzym trawienny trypsyna przecinający łańcuch peptydowy w pobliżu zasadowych aminokwasów) lub ograniczać się wyłącznie do rozpoznawania typu wiązania chemicznego (np. niektóre dehydrogenazy alkoholowe utleniają specyficznie alkohole pierwszorzędowe do aldehydów). Enzymy potrafią również rozpoznawać przestrzenną strukturę cząsteczek chemicznych, dzięki czemu potrafią być specyficzne względem tak zwanych izomerów optycznych (enancjomerów) – czyli cząsteczek niemal identycznych, różniących się tylko przestrzenną organizacją podstawników wokół tzw. centrów stereogenicznych (np. chiralnych atomów węgla – chiralne cząsteczki, tak jak nasze ręce, które wyglądają jak swoje lustrzane odbicia). Dzięki enancjospecyficzności enzymy są niezwykle przydatne szczególnie w przemyśle farmaceutycznym, gdzie ich zdolność do produkcji chiralnych czystych enancjomerycznie leków może decydować o tym, czy produkt będzie miał zbawienne działanie lecznicze, czy też stanie się zagrażającą życiu trucizną [7, 14]. Co więcej zastosowanie enancjoselektywnych biokatalizatorów w syntezie ma też duże znaczenie ekonomiczne, gdyż lek jest syntetyzowany tylko w aktywnej formie. Dla każdego centrum stereogenicznego zyskujemy więc dwa razy więcej substancji aktywnej, podczas gdy w przypadku syntezy nie-enancjoselektywnej otrzymujemy tylko połowę możliwej ilości produktu. Mając czysty optycznie lek firma farmaceutyczna nie musi też przeprowadzać kosztownych badań klinicznych skierowanych na wykrycie efektów ubocznych, potencjalnie powodowanych przez nieaktywny klinicznie enanjomer leku. Nie ma też potrzeby przeprowadzenia kosztownego oczyszczania substancji aktywnej z niepotrzebnego enancjomeru, jeżeli jego podanie powoduje niepożądane skutki.
Bardzo często aktywność katalityczna enzymów podlega ścisłej kontroli i regulacji. Dobrym przykładem takiego zachowania są enzymy trawienne, katalizujące rozkład białek. Enzymy te wytwarzane są m.in. w gruczołach ściany żołądka. Gruczoły ściany żołądka w dużej mierze zbudowane są z białek, więc enzymy trawienne muszą być początkowo nieaktywne (wydzielane w formie tzw. proenzymu pepsynogenu), żeby nie trawiły własnego macierzystego gruczołu. I dopiero wtedy, gdy już wydzielą się do żołądka, gdzie panuje niskie pH (obecny jest tam kwas żołądkowy), ulegają samo-aktywacji (autokatalitycznie same odcinają swój niewielki fragment) i stają się bardzo aktywnymi katalizatorami ułatwiającymi trawienie białek (pepsyna) [5]. Dodatkowo nasz żołądek wyścielony jest specjalnymi komórkami odpornymi na działanie pepsyny – inaczej sami moglibyśmy siebie strawić po dobrym obiedzie! Co ciekawe inne enzymy trawienne, wytwarzane w trzustce (np. chymotrypsyna), a działające w jelicie cienkim, gdzie panuje pH zasadowe, trawią białka tylko i wyłącznie w warunkach wysokiego pH. Tym samym ich aktywność enzymatyczna jest precyzyjnie przystosowana do miejsca, w którym mają za zadanie działać.
Aktywność enzymatyczna (czyli zdolność do przyspieszania reakcji) podlega dynamicznej regulacji. Zanim zajdzie reakcja, najpierw musi powstać niekowalencyjne połączenie między enzymem (E) i substratem (S), czyli tzw. kompleks ES. Ponieważ sama reakcja chemiczna jest bardzo często wolniejsza od procesu powstawania i rozpadu kompleksu ES, szybkość katalizowanej przez enzym reakcji zależy nieliniowo od stężenia substratu i ma kształt hiperboli. Oznacza to, że w miarę zwiększania się stężenia substratu szybkość reakcji tylko początkowo gwałtownie rośnie po czym przyrost szybkości stopniowo zwalnia, aż osiąga wysycenie (Ryc. 3).

Ryc. 3. Wykres zależności szybkości reakcji enzymatycznej V od stężenia substratu [S], gdzie Vmax – maksymalna szybkość reakcji enzymatycznej, osiągana dla nieskończenie wysokiego stężenia substratu. Km stała Michaelisa oznaczająca stężenie substratu, przy jakim szybkość reakcji katalizowanej przez enzym osiąga połowę Vmax.
Zrozumienie tego efektu było możliwe dzięki badaniom trójki uczonych: francuskiego chemika Victora Henriego, niemieckiego biochemika Leonora Michaelisa i kanadyjskiej doktor medycyny Maud Menten. To oni sformułowali najsłynniejsze w biochemii równanie Michaelisa-Menten, będące podstawowym prawem opisującym zachowanie enzymów [8].
Co ciekawe wiele enzymów zmienia swoją aktywność w zależności od stężenia substratu i innych substancji w sposób jeszcze bardziej złożony. Częstym zjawiskiem jest spowolnienie reakcji gdy powstaje dużo produktu. W ten sposób organizm zabezpiecza się przed akumulacją zbyt dużej ilości substancji chemicznych, które w wysokim stężeniu mógłby wywrzeć toksyczny efekt na komórkę. Efekt taki nazywany jest inhibicją produktową, jeżeli produkt wiąże się w centrum aktywnym, lub ujemnym efektem allosterycznym, jeżeli miejsce wiązania się inhibitora-produktu znajduje się w innej części enzymu.

Ryc. 4. Schematyczne przedstawienie różnych sposobów regulacji aktywności enzymatycznej przez inhibitory. A) Normalna reakcja enzymatyczna, w której enzym wiąże substrat i po reakcji uwalnia produkt, B) Inhibitor kompetycyjny wiąże się w centrum aktywnym uniemożliwiając związanie substratu i zajście reakcji, C) Związanie się z enzymem czynnika allosterycznego wpływa na zmianę struktury enzymu, co pośrednio oddziaływuje na geometrię centrum aktywnego. W rezultacie enzym nie wiąże skutecznie substratu i reakcja nie zachodzi.
Aktywność enzymatyczna może podlegać jeszcze innej regulacji, najczęściej (choć nie jedynie) gdy enzym składa się z kilku jednakowych pod względem składu aminokwasowego podjednostek białkowych, z których każda zawiera centrum aktywne. Mówimy wtedy, że enzym ma budowę multimeryczną (np. jeżeli składa się z czterech identycznych podjednostek, jest homotetramerem – Ryc. 5).
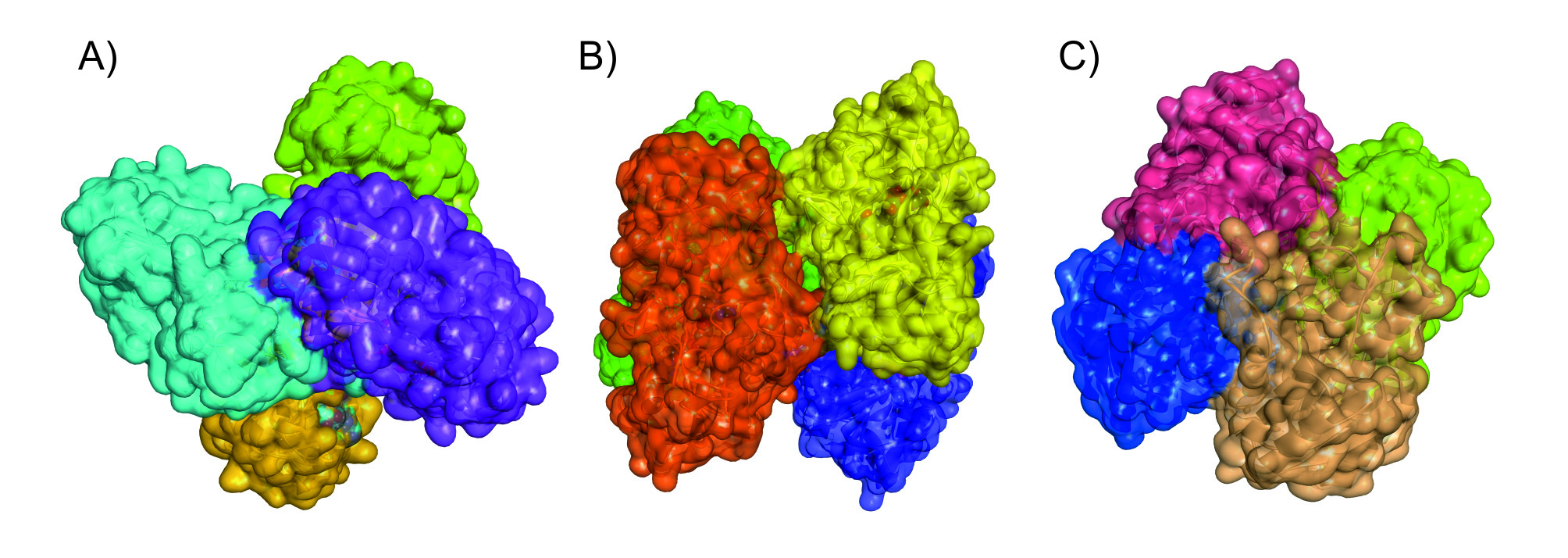
Ryc. 5. Przykłady białek tetramerycznych: A) dehydrogenaza semialdehydu aspartaniowego z Trichophyton rubrum [10], B) Δ1-dehydrogenaza 3-ketosteroidowa z Rhodococcus erythropolis [16], C) dehydrogenaza (S)-1-fenyloetanolowa z Aromatoleum aromaticum [6].
Bardzo często takie enzymy działają kooperatywnie – tzn. związanie w jednej z podjednostek substratu powoduje zmianę geometrii białka. Zmiana kształtu podjednostki zawierającej substrat pociąga za sobą przemieszczenie położenia aminokwasów na styku pomiędzy podjednostkami. W efekcie następuje również zmiana geometrii pozostałych podjednostek (tzw. zmiana konformacji), które niejako „czują”, że ich sąsiad związał pierwszą cząsteczkę substratu. W rezultacie ich zdolność do wiązania cząsteczek substratu w centrach aktywnych (a więc termodynamiczne prawdopodobieństwo powstania kompleksu ES) może wzrosnąć (dodatni efekt kooperatywny) lub zmaleć (ujemny efekt kooperatywny) [9, 12]. W ten sposób ewolucyjnie zaprogramowany efekt regulacji pozwala organizmowi dostosować efektywność katalizatorów do zmieniających się warunków zewnętrznych (np. zmiennego stężenia substancji odżywczych).
Białkowa budowa enzymów
Jak już wspomniano, enzymy mają bardzo skomplikowaną budowę. Ponieważ są białkami, zbudowane są z aminokwasów. Większość organizmów wyższych (do których i my się zaliczamy) wykorzystuje tylko 21 różnych aminokwasów, które niczym klocki lego połączone są w długie łańcuchy za pomocą tzw. wiązania peptydowego (Ryc. 6).

Ryc. 6. Struktura wiązania peptydowego (szary romb) pomiędzy dwoma aminokwasami alaniny. Dzięki sprzężeniu elektronowemu wolnej pary elektronowej atomu azotu i układu elektronów grupy karbonylowej wiązanie peptydowe jest płaskie. Ułożenie aminokwasów w łańcuchu polipeptydowym to struktura I-rzędowa.
To, w jakiej kolejności poukładane są różne aminokwasy w danym białku jest zapisane w kodzie genetycznym (DNA) i w jeszcze nie do końca zrozumiały dla nas sposób niesie informację nie tylko o kolejności (tak zwanej sekwencji) aminokwasów w łańcuchu, ale również o całkowitym trójwymiarowym kształcie białka. Okazuje się bowiem, że dojrzałe białko (czyli np. jakiś enzym) przypomina bardziej kłębek włóczki, którym bawił się młody kociak, niż prostą wstążkę. Nić aminokwasowa przyjmuje skomplikowaną i uporządkowaną strukturę spirali (zwaną helisą) lub płaszczyzny (zwaną β-kartką) (Ryc, 7A, B). Helisy i β-kartki będące strukturą II-rzędową białek tworzą z kolei przestrzenne struktury III-rzędowe, które w formie przypominają nie tylko kłębek włóczki lecz także i fantazyjne pierścienie czy też beczki (Ryc. 4B, C).

Ryc. 7. Hierarchiczna struktura białek tworzona przez sekwencję aminokwasową łańcucha polipeptydowego (struktura 1o-rzędowa) gdzie A) to sposób zwinięcia (struktura 2o-rzędowa) w charakterystyczne helisy (czerwone), β-kartki (niebieskie) i spinki (zielone), B) i C) przestrzenna organizacja struktur 2o-rzędowych w strukturę 3o-rzędową zawierającą charakterystyczne motywy takie jak pęki helis, zwijającą się β-kartkę, β-baryłkę, których funkcją może być wytworzenie miejsca wiązania cząsteczki (ciemno i jasno-szara kulkowa struktura substratu w B) oraz zapewnienie wyizolowanego środowiska dla aktywnych aminokwasów katalizujących reakcję (aminokwasy w centrum baryłki w C). Na panelu D) przedstawiono 4o-rzędową organizację tworzoną przez odrębne łańcuchy polipeptydowe (oznaczone różnymi kolorami); A) fragment struktury modelu homologicznego Δ1-dehydrogenazy 3-ketosteroidowej ze Sterolibacterium denitrificans [3], B) komórkowe białko wiążące retinol z Homo sapiens (kod PDB 5FAZ) C) i D) dehydrogenaza tiocyjanianowa z Thioalkalivibrio paradoxus (kod PDB: 5F75).
Niektóre enzymy są naprawdę wielkimi cząsteczkami chemicznymi, składającymi się z wielu splątanych ze sobą łańcuchów i dziesiątków tysięcy atomów. Tym bardziej jest dla nas niezrozumiałe w jaki sposób białka za każdym razem, zazwyczaj bezbłędnie i w większości wypadków zupełnie samoistnie, zwijają się w te skomplikowane struktury. Pomimo wielu badań w tej dziedzinie oraz ogromnego postępu metod symulacji komputerowych zwijania się (tzw. fałdowania) białek wciąż nie jesteśmy w stanie przewidzieć z wystarczająca dokładnością przestrzennej struktury białek bazując wyłącznie na sekwencji aminokwasowej. To właśnie z tego powodu co roku (od 1994) odbywa się swego rodzaju eksperyment: światowe mistrzostwa w przewidywaniu struktur białkowych CASP (Critical Assessment of protein Structure Prediction) [13], w ramach których ponad 100 grup naukowców rywalizuje w przewidywaniu struktury białka o niedawno poznanej, ale wciąż nie opublikowanej strukturze.
Należy sobie jednak postawić pytanie, czemu ma służyć tak skomplikowana budowa białek i enzymów? Enzymolodzy byli bardzo zaskoczeni gdy odkryli, że niejednokrotnie w samym procesie przyspieszania reakcji bierze udział tylko kilka aminokwasów! Sama reakcja chemiczna zachodzi bowiem w tak zwanym „centrum aktywnym”, które jest najczęściej niewielkim wgłębieniem lub niszą w enzymie, do którego wiążą się cząsteczki chemiczne (substraty) przetwarzane przez enzym. To właśnie w stronę przestrzeni centrum aktywnego zwrócone są w precyzyjnie określonych miejscach aminokwasy zaangażowane w przyspieszenie reakcji chemicznej. Centrum aktywne można więc wyobrazić sobie jak najeżoną wyspecjalizowanymi ramionami robotów taśmę produkcyjną w wielkie fabryce. Od dziesięcioleci próbujemy zrozumieć, jak enzymy osiągają te fenomenalne rezultaty za pomocą relatywnie niewielkich i uniwersalnych środków. Niestety wciąż poszukujemy pełnej odpowiedzi, bo czynników odpowiedzialnych za katalityczną perfekcję enzymów wydaje się być wiele. Rozumiemy, że kluczem do wszystkiego jest właśnie obniżanie energii aktywacji danej reakcji za pomocą tych kilku odpowiednio ulokowanych aminokwasów lub kofaktorów, czyli niewielkich cząsteczek organicznych lub kompleksów metali, które poszerzają spektrum dostępnych chemicznych procesów. Enzymów jest jednak tak wiele i tak wiele różnych reakcji potrafią przyspieszać, że enzymolodzy mają wiele pracy na kolejne dziesiątki lat badań. W samym ludzkim organizmie ocenia się – na podstawie analizy genomu – że biokatalizatorów, katalizujących prawie 900 różnych procesów, może być ponad 2 700 [17]. A większość organizmów żywych naszej planety nie została jeszcze scharakteryzowana genetycznie!
Centrum aktywne
Należy sobie zadać pytanie dlaczego Natura wytworzyła tak skomplikowane cząsteczki, by ostatecznie zbudować coś tak prostego, jak centrum aktywne? Czy enzymy nie mogłyby składać się właściwe tylko z tych kilku kluczowych aminokwasów, które katalizują reakcję? Przecież tak właśnie budujemy nasze katalizatory my, chemicy – i jakoś to działa! Powodów, dla których Natura wybrała to pozornie bardziej skomplikowane rozwiązanie jest kilka. Po pierwsze, jak już wspomniałem, białka, a w szczególności enzymy, produkują się poniekąd same. Po przetłumaczeniu kodu genetycznego na łańcuch białkowy, sama struktura polimeru białkowego zwija się w docelowy kształt. Ten kształt jest tak zaprojektowany przez ewolucję, aby umieścić katalitycznie aktywne aminokwasy w odpowiedniej, optymalnej pozycji. Pozostałe aminokwasy nie tylko stanowią rusztowanie, na którym katalitycznie aktywne aminokwasy są zamocowane, ale również budują kształt centrum aktywnego, które przypomina formę pod odlew rzeźby dla reagujących w reakcji cząsteczek. Innymi słowy cząsteczki reagentów „pasują”, do centrum aktywnego niczym ręka do dobrze dobranej rękawiczki i każdy fragment cząsteczki chemicznej wchodzi do odpowiedniego „palca” tej rękawiczki. Oczywiście dobre „dopasowanie” reagenta do centrum aktywnego wykracza poza czysto makroskopowe mechaniczne dopasowanie kształtu, chociaż jest to również istotny czynnik (są to tak zwane efekty steryczne) (Ryc. 8).
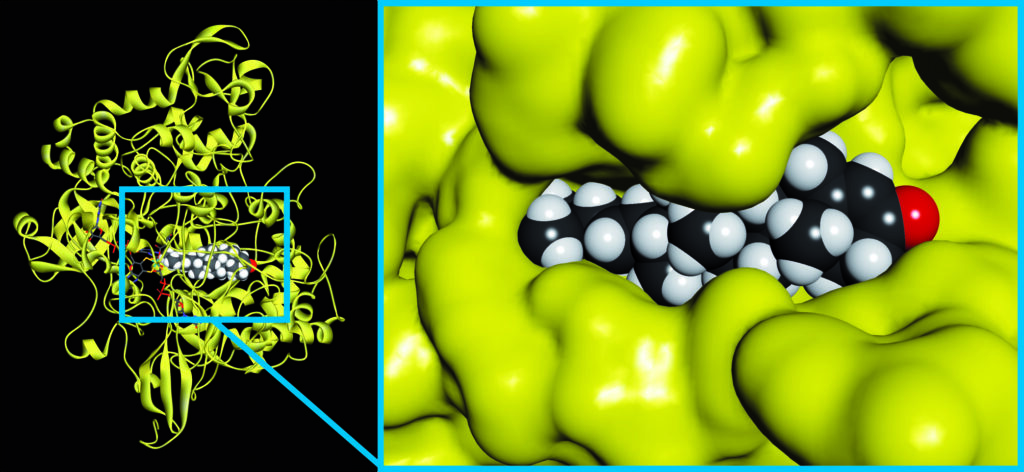
Ryc. 8. Katalityczna podjednostka dehydrogenazy C25 steroidowej wraz z substratem w jej centrum aktywnym [18]. Na prawym panelu przedstawiono doskonałe dopasowanie substratu do powierzchni białka. Część aminokwasów przesłaniających substrat od strony czytelnika ukryto, w celu lepszej wizualizacji kompleksu enzym-substrat (ES).
Bardzo ważną rolę pełnią też oddziaływania elektrostatyczne – aminokwasy o naładowanych (dodatnio lub ujemnie) łańcuchach bocznych umieszczone są w takich miejscach, aby korzystnie oddziaływać z przeciwnie naładowanymi grupami substratu lub też umożliwić powstanie stabilizujących kompleks enzymu z substratem wiązań wodorowych. To właśnie dzięki tym doskonale zaprojektowanym oddziaływaniom centrum aktywnego z substratem enzymy uzyskują niezwykłą przewagę nad naszymi prostymi chemicznymi katalizatorami. Przewagą tą jest wspomniana wcześniej selektywność. Rozpoznając kształty i właściwości cząsteczek chemicznych nasza „enzymatyczna rękawica” dba nie tylko o to aby „kciuk” czy „palec wskazujący” cząsteczki trafił w odpowiednie miejsce rękawicy, ale jest też w stanie rozróżnić chemiczną „malutką rączkę dziecka”, od „wielkiej męskiej graby”, czy „kobiecej rączki o długich palcach i wypielęgnowanych paznokciach”. Innymi słowy enzym wybierze z wielu cząsteczek chemicznych pływających w komórkowej cytoplazmie tylko te związki, do których modyfikacji został zaprojektowany. Dzięki tej niezwykłej zdolności w naszych komórkach potrafią biec obok siebie bardzo różne reakcje chemiczne, podczas gdy my, w chemicznej fabryce, musimy każdy z procesów chemicznych prowadzić w osobnym reaktorze, by nasze katalizatory „nie pomyliły się” i modyfikowały tylko jeden wybrany przez nas związek chemiczny.
Selektywność jest wielką zaletą enzymów i ma bardzo duże znaczenie dla ich zastosowania w przemyśle. Co ciekawe – jest jednocześnie zaletą i wadą biokatalizatorów. Otóż tak długo, jak chcemy odtworzyć w fabryce proces, który występuje w Naturze (na przykład chcemy syntetyzować antybiotyk wykorzystując enzymy z grzybów, które naturalnie go wytwarzają), tak długo ta niezwykła selektywność enzymów działa na naszą korzyść. Enzym wytworzy tylko jeden, pożądany przez nas produkt i kompletnie zignoruje zanieczyszczenia. W przemyśle jednak bardzo często chcemy stworzyć cząsteczki, których Natura nie używa albo które zawierają nietypowe atomy. Wtedy ta wysoka specjalizacja biokatalizatorów działa na naszą niekorzyść, bo enzymy są zbyt wysoce wyspecjalizowane, by dobrze przyspieszać przemianę nietypowych cząsteczek. Nietypowe cząsteczki mające trochę inny chemiczny kształt oraz rozkład ładunków i nie pasują dobrze do centrum aktywnego. Aby poradzić sobie z tym problemem biotechnolodzy „przesiewają” wiele enzymów, pochodzących z różnych organizmów, by znaleźć takie, które nie są aż tak bardzo wyspecjalizowane lub posiadają inną, bardziej pasująca do celów danej syntezy, specyficzność substratową. Czasem udaje się znaleźć enzym, którego ewolucja nie doprowadziła jeszcze do skrajnej specjalizacji. Dzięki temu jest on w stanie rozpoznać i związać w centrum aktywnym szerszą gamę związków chemicznych i może służyć naszym celom. Takie poszukiwania przypominają jednak szukanie igły w stogu siana – nigdy nie mamy pewności, że odnajdziemy enzym o pożądanych cechach. Na szczęście dzięki postępom biologii molekularnej nie jesteśmy zdani tylko na łut częścią i Naturę – możemy liczyć na pomoc ze strony przyspieszonej i ukierunkowanej ewolucji enzymów w laboratorium.
Sztuczna ewolucja enzymów
Proces ewolucji polega na stopniowych, losowych zmianach w kodzie genetycznym kodującym sekwencje białek. Enzymy odpowiedzialne za kopiowanie DNA są zazwyczaj bardzo dokładne i bardzo rzadko popełniają błędy. Czasem jednak pojedyncze pomyłki „umkną ich uwadze” i w ten sposób następuje mutacja punktowa w kodzie DNA. Niekiedy zmiana pojedynczego elementu DNA nie prowadzi do poważnych zmian, czasem zmodyfikowane białko ma własności mniej przydatne niż jego pierwowzór, ale czasem jest ono lepsze. Ponieważ lepsze rozwiązania zwiększają szanse na przeżycie danego organizmu i wydanie na świat potomstwa (a wiec przekazanie nowego typu białka swoim dzieciom) dobre rozwiązania się kumulują, a złe eliminują (bo często zmniejszają szansę przeżycia i przekazania gorszego białka potomstwu).
W sztucznie kierowanej ewolucji to my decydujemy, jakie będą warunki „przeżycia” danej losowej mutacji. Są nim zdolności enzymu do katalizowania wybranej przez nas reakcji chemicznej. Dzięki postępom w dziedzinie biologii molekularnej posiadamy specjalne narzędzia biotechnologiczne umożliwiające częste wprowadzanie losowych jak i celowych zmian w DNA i następnie produkcji białek kodowanych przez zmienione DNA w bakteriach. Posiadamy też narzędzia umożliwiające mieszanie się kodu genetycznego pomiędzy podobnymi, homologicznymi genami, w podobny sposób jak w procesie płciowego rozmnażania [19]. Powtarzając proces mutacji i selekcji wielokrotnie dla dużej liczby wariantów enzymu (niczym kolejne pokolenia w normalnej ewolucji) jesteśmy w stanie przekształcić enzym, który był słaby, w katalizowaniu nieznanej Naturze reakcji, w super katalizator, który nie tylko przyspieszy reakcję nawet i 1000-krotnie, ale jeszcze zniesie wysokie stężenie rozpuszczalników organicznych, wysoką temperaturę czy jakże odmienne od przytulnego wnętrza komórki warunki panujące w reaktorze chemicznym. Dzięki poszerzającej się wiedzy na temat zależności struktury białek i ich właściwości jesteśmy coraz częściej w stanie przewidywać (za pomocą modelowania molekularnego), jakie modyfikacje należy wprowadzać, by skierować ewolucję w danym kierunku, i jakie rejony białka najlepiej jest poddawać mutacjom. Dlatego nie musimy już poruszać się zupełnie na ślepo, co pozwala optymalizować właściwości katalityczne enzymów metodą ewolucji kierowanej dla mniejsze ilości białek [11]. Co więcej dzięki technikom inżynierii genetycznej i mikrobiologii jesteśmy w stanie wytwarzać te enzymy w sposób tani i bezpieczny dla środowiska. Stało się to możliwe dzięki zrozumieniu kodu genetycznego, stanowiącego instrukcję do produkcji poszczególnych enzymów oraz rozwojowi narzędzi molekularnych pozwalających tę instrukcję kopiować, modyfikować i przenosić do innych organizmów. Za pomocą technik transformacji genetycznych wprowadzamy odpowiednio przygotowaną genetyczną instrukcję do pospolitych mikroorganizmów (np. bakterii Escherichii coli czy drożdży Pichia pastoris), namnażamy je i wydajemy im „chemiczny rozkaz” nakazujący produkcję pożądanego enzymu w dużej ilości. Ten „chemiczny rozkaz”, nazywamy fachowo „indukcją”, wydajemy za pomocą dodatku do hodowli chemicznych związków (takich jak np. laktoza czy ramnoza), które zmieniają właściwości białek regulujących ekspresję genetyczną w mikroorganizmach. W obecności tych substancji białka regulatorowe odczepiają się od DNA mikroorganizmu (w szczególności zaś od DNA wprowadzonego przez biologów molekularnych sztucznie do zmodyfikowanej bakterii) i umożliwiają transkrypcję zakodowanej instrukcji produkcji pożądanego enzymu. Dzięki takim osiągnięciom genetycznie zmodyfikowane organizmy (czyli GMO) dostarczają nam m.in. tak zwanych rekombinowanych enzymów. Jest to sposób produkcji podpuszczki do produkcji serów (nie musimy więc zabijać jagniąt by ekstrahować enzym z ich żołądków), wytwarzania enzymów rozkładających skrobię do syropu glukozowego (soki słodkie nawet w zimie), czy produkujących lipazy usuwające tłuste plamy z pranej odzieży (tanie proszki zawierające enzymy pozwalają na pranie w niskiej temperaturze, co oszczędza prąd i chroni środowisko). Na marginesie należy wspomnieć, że te same metody inżynierii genetycznej służą do produkcji bardzo cennych leków, takich jak insulina ludzka,ludzki hormon wzrostu czy też innowacyjne leki przeciw chorobom autoimmunologicznym takim jak choroba Leśniowskiego-Crohna, wrzodziejące zapalenie jelita grubego, łuszczyca czy reumatoidalne zapalenie stawów [2]. Te biologiczne leki również zbudowane są z aminokwasów i wytwarzane są w zmodyfikowanych genetycznie komórkach eukariotycznych (tj. nie bakteryjnych – najczęściej w komórkach jajnika chomika chińskiego – CHO). Przeciwciała wytwarzane w GMO mają również zastosowanie w diagnostyce biomedycznej (jako molekularne czujniki wykrywają, np. hCG w teście ciążowym) jak również jako sztuczne biokatalizatory, czyli rodzaj wytworzonych przez chemików enzymów [20]. Jak więc widać nie należy się ślepo obawiać GMO dla samej zasady – to co modyfikowane genetycznie niejednokrotnie ratuje nasze życie i zdrowie, a przy tym wspiera ochronę środowiska.
Podsumowanie
Otaczający nas świat zmienia się czasem niepostrzeżenie. Jeszcze 20-30 lat temu enzymy wykorzystywali tylko specjaliści w laboratoriach czy fabrykach farmaceutycznych. Dziś, czasem nieświadomie, wykorzystują je gospodynie domowe (lub samodzielnie robiący pranie mężczyźni) dolewając żelu do prania z formułą enzymatyczną usuwającą plamy z białek, tłuszczu i cukrów. Korzystają z nich piekarze, dodający enzymatycznych polepszaczy, które sprawiają, że chleb jest bardziej sprężysty i aromatyczny, czy kucharze, wykorzystujący enzymatyczne marynaty, by mięso nabrało odpowiedniej miękkości i delikatności. Stosuje się enzymatyczne preparaty proteolityczne, by sklarować piwo produkowane w lokalnym mikro-browarze. No i oczywiście każdy z nas, w każdej sekundzie naszego istnienia, wykorzystuje tysiące enzymów – po to by po prostu żyć.
Literatura
- Arrhenius, S, Über die Dissociationswärme und den Einfluss der Temperatur auf den Dissociationsgrad der Elektrolyte. In Zeitschrift für Physikalische Chemie, 1889; Vol. 4, p 96.
- Brekke, OH; Sandlie, I. (2003). Therapeutic antibodies for human diseases at the dawn of the twenty-first century. Nat. Rev. Drug Discov. 2: 52-62.
- Chiang, YR; Ismail, W; Gallien, S; Heintz, D; Van Dorsselaer, A; Fuchs, G. (2008). Cholest-4-En-3-one-Delta(1)-dehydrogenase, a flavoprotein catalyzing the second step in anoxic cholesterol metabolism. Appl. Environ. Microbiol. 74: 107-113.
- Glasstone, SLKJEH, The Theory of Rate Processes The Kinetics of Chemical Reactions, Viscosity, Diffusion and Electrochemical Phenomena. Mcgraw-Hill Book Compagny: New York, N.Y., 1941.
- Herriott, RM. (1938). Kinetics of the Formation of Pepsin from Swine Pepsinogen and Identification of an Intermediate Compound. J. Gen. Physiol. 22: 65-78.
- Hoffken, HW; Duong, M; Friedrich, T; Breuer, M; Hauer, B; Reinhardt, R; Rabus, R; Heider, J. (2006). Crystal structure and enzyme kinetics of the (S)-specific 1-phenylethanol dehydrogenase of the denitrifying bacterium strain EbN1. Biochemistry 45: 82-93.
- Izake, EL. (2007). Chiral discrimination and enantioselective analysis of drugs: an overview. J. Pharm. Sci. 96: 1659-76.
- Johnson, KA; Goody, RS. (2011). The Original Michaelis Constant: Translation of the 1913 Michaelis–Menten Paper. Biochemistry 50: 8264-8269.
- Koshland, DE; Némethy, G; Filmer, D. (1966). Comparison of Experimental Binding Data and Theoretical Models in Proteins Containing Subunits*. Biochemistry 5: 365-385.
- Li, Q; Mu, Z; Zhao, R; Dahal, G; Viola, RE; Liu, T; Jin, Q; Cui, S. (2016). Structural Insights into the Tetrameric State of Aspartate-β-semialdehyde Dehydrogenases from Fungal Species. Scientific Reports 6: 21067.
- Lutz, S. (2010). Beyond directed evolution—semi-rational protein engineering and design. Curr. Opin. Biotechnol. 21: 734-743.
- Monod, J; Wyman, J; Changeux, JP. (1965). On the Nature of Allosteric Transitions: A Plausible Model. J. Mol. Biol. 12: 88-118.
- Moult, J; Pedersen, JT; Judson, R; Fidelis, K. (1995). A large-scale experiment to assess protein structure prediction methods. Proteins: Struct. Funct. Bioinform. 23: ii-v.
- Nguyen, LA; He, H; Pham-Huy, C. (2006). Chiral drugs: an overview. Int. J. Biomed. Sci. 2: 85-100.
- Pauling, L. (1948). Chemical achievement and hope for the future. Am. Sci. 36: 51-8.
- Rohman, A; van Oosterwijk, N; Thunnissen, A-MWH; Dijkstra, BW. (2013). Crystal Structure and Site-directed Mutagenesis of 3-Ketosteroid Δ1-Dehydrogenase from Rhodococcus erythropolis SQ1 Explain Its Catalytic Mechanism. J. Biol. Chem. 288: 35559-35568.
- Romero, P; Wagg, J; Green, ML; Kaiser, D; Krummenacker, M; Karp, PD. (2004). Computational prediction of human metabolic pathways from the complete human genome. Genome Biology 6: R2.
- Rugor, A; Wójcik-Augustyn, A; Niedzialkowska, E; Mordalski, S; Staroń, J; Bojarski, A; Szaleniec, M. (2017). Reaction mechanism of sterol hydroxylation by steroid C25 dehydrogenase – Homology model, reactivity and isoenzymatic diversity. J. Inorg. Biochem. 173 28–43
- Stemmer, WPC. (1994). Rapid evolution of a protein in vitro by DNA shuffling. Nature 370: 389-391.
- Wentworth, P; Janda, KD. (2001). Catalytic antibodies. Cell. Biochem. Biophys. 35: 63-87.